

Abstract
Myeloid dysplastic syndrome (MDS) reflects a preleukemic bone marrow (BM) disorder with limited treatment options and poor disease survival. As only a minority of MDS patients are eligible for curative hematopoietic stem cell transplantation, there is an urgent need to develop alternative treatment options. Chronic activation of Wnt/β-catenin has been implicated to underlie MDS formation and recently assigned to drive MDS transformation to acute myeloid leukemia. Wnt/β-catenin signaling therefore may harbor a pharmaceutical target to treat MDS and/or prevent leukemia formation. However, targeting the Wnt/β-catenin pathway will also affect healthy hematopoiesis in MDS patients. The control of Wnt/β-catenin in healthy hematopoiesis is poorly understood. Whereas Wnt/β-catenin is dispensable for steady-state erythropoiesis, its activity is essential for stress erythropoiesis in response to BM injury and anemia. Manipulation of Wnt/β-catenin signaling in MDS may therefore deregulate stress erythropoiesis and even increase anemia severity. Here, we provide a comprehensive overview of the most recent and established insights in the field to acquire more insight into the control of Wnt/β-catenin signaling in healthy and inefficient erythropoiesis as seen in MDS.
Introduction
Wnt/β-catenin signaling underlies the control on tissue homeostasis by instructing stem cell self-renewal and differentiation.1-4 Binding of canonical Wnt ligands with the heterodimeric Wnt receptor complex stimulates canonical Wnt signaling by inactivating the β-catenin destruction complex.2 Stabilization of β-catenin accordingly allows nuclear translocation and gene regulation by β-catenin to instruct cell behavior.1-3 Unrestrained activation of Wnt/β-catenin signaling has been implicated to underlie myeloid dysplastic syndrome (MDS) development.5-10 MDS represents a preleukemic condition characterized by clonal outgrowth of malignant hematopoietic stem cells (HSCs) and anemia with poor overall survival.11-13 Treatment options of MDS are limited, as only a minority of MDS patients are eligible for curative HSC transplantation.11-13
Recently, chronic activation of β-catenin also has been assigned to drive transformation of MDS to acute myeloid leukemia (AML).8,14 Manipulation of β-catenin signaling may therefore offer a therapeutic treatment strategy to prevent or delay leukemia formation in MDS patients. However, manipulation of β-catenin signaling will also affect the healthy hematopoietic compartment in MDS.28 The control of Wnt/β-catenin signaling in healthy hematopoiesis is complex.16-24 Wnt/β-catenin signaling is dispensable for steady-state erythropoiesis but required to promote stress erythropoiesis in response to bone marrow (BM) injury and anemia.25,26 Redirecting β-catenin signaling in MDS to prevent or delay AML formation may therefore also deregulate stress erythropoiesis and even increase anemia severity. In order to acquire more insight in the regulation of Wnt/β-catenin signaling on both homeostatic and inefficient red blood cell (RBC) formation, we here provide a comprehensive review of the most recent and established insights concerning Wnt/β-catenin signaling during healthy erythropoiesis and MDS.
Wnt/β-catenin signaling secures initiation of erythropoiesis during embryogenesis
Wnt/β-catenin signaling is well known to control embryogenesis, as initially discovered in wingless Drosophila mutants that developed wing-to-notum transformation upon expression of Wnt mutants.27 Wnt/β-catenin signaling dictates embryogenesis by controlling self-renewal and fate decision of embryonic stem (ES) and progenitor cells.26 The earliest form of erythropoiesis during embryogenesis, known as primitive erythropoiesis, is partly controlled by Wnt/β-catenin signaling.28,29 Primitive RBCs develop from “blood islands” of the yolk sac to secure oxygen transport and promote vascular remodeling during early embryonic development.30 Failure to induce primitive erythropoiesis is uniformly associated with embryonic lethality, overall underscoring the critical role of primitive RBCs in the development of the rapidly growing embryo.31 Primitive erythroid progenitors that arise from the yolk sac, also known as EryP-CFC (primitive erythroid progenitor colony-forming cells), express several Wnt pathway components such as β-catenin, the Wnt receptor Fzd7, and the Wnt ligand coactivator Rspo3 that decrease during differentiation toward primitive RBCs.28,29 Development of this early wave of primitive erythropoiesis coincides with activation of Wnt/β-catenin signaling, as revealed by GFP expression in blood islands in the yolk sac of TCF/Lef;H2B-GFP Wnt reporter mouse embryos.28 This suggests Wnt/β-catenin signaling to stimulate development of EryP-CFCs and/or their expansion. A notion that is also underscored by the ability of Wnt3a to expand Ery-CFCs in isolated yolk sacs in vitro.32 Moreover, forced activation of Wnt/β-catenin signaling in mouse ES cell cultures also reveals canonical Wnt signaling to stimulate ES cell differentiation to primitive erythroid lineage formation.21 Overall, this indicates Wnt/β-catenin signaling to stimulate the onset of primitive erythropoiesis.
Completion of the first wave of primitive erythropoiesis coincides with initiation of a second transient wave of erythropoiesis.33,34 This second wave is also initiated in the yolk sac and drives formation of definitive erythroid progenitors called BFU-E (burst-forming units-erythroid) that travel through the bloodstream and colonize the fetal liver.33,34 Wnt3a stimulation can expand these erythroid progenitors in isolated yolk sac in vitro.32 The second wave of embryonic erythropoiesis secures formation of sufficient definitive RBCs during the onset of the final wave of embryonic erythropoiesis.33,34 The final wave of embryonic erythropoiesis arises from the major embryonic endothelial arteries between the gonad and mesonephros (AGM) region during a process called endothelial-to-hematopoietic transition (EHT).35 Hematopoietic stem and progenitor cells (HSPCs) arising from EHT expand in the fetal liver and ultimately home to the BM, which thereby replaces RBC production by the fetal liver.36 Wnt/β-catenin signaling is required to promote HSC formation, as somatic loss of β-catenin in the embryonic endothelium severely impairs EHT at the AGM in the VEC-Cre;Ctnnb1F/F;R26R-YFP transgenic mouse model.17,37 Wnt3a expression is also tightly regulated in the AGM and peaks at EHT initiation.38 Whereas loss of Wnt3a in Wnt3a−/− mouse embryos severely reduces EHT driven generation of HSPCs, the absence of Wnt3a does not completely abolish EHT, nor does it affect subsequent erythropoiesis in the fetal liver.39 This indicates that Wnt/β-catenin signaling may also be controlled by alternative canonical Wnt ligands to secure EHT progression, such as Wnt10b, which is also expressed during development of definitive hematopoiesis.40
The control of Wnt/β-catenin signaling on initiation of erythropoiesis during mouse embryogenesis is also reported for Xenopus.37 Initiation of primitive hematopoiesis in Xenopus is marked by a peak in Wnt/β-catenin signaling as revealed in canonical Wnt reporter pbin8Lef;dGFP Xenopus tropicalis embryos.37 In Xenopus embryos, primitive hematopoiesis arises from ventral blood islands, recognized as the equivalent of the mammalian blood islands in the yolk sac. Wnt signaling during initiation of primitive hematopoiesis in Xenopus seems completely dependent on Wnt4, as Wnt4 morpholino crossed with pbin8Lef;dGFP largely precludes expression of hematopoietic cell markers SCL and T3 globin.37 Wnt4-driven signaling is however less understood compared with Wnt3a. In contrast to Wnt3a, Wnt4 is able to also promote non-canonical Wnt signaling.41 Loss of Wnt4 in Xenopus therefore may hamper primitive erythropoiesis by hampering both Wnt/β-catenin and noncanonical Wnt signaling. Noncanonical Wnt signaling represents multiple pathways that instead of β-catenin use alternative secondary messengers.42 Noncanonical Wnt signaling promotes HSC quiescence in mouse BM and self-renewal of mouse-derived HSPCs in vitro.43,44 Although noncanonical signaling has not been reported to control erythropoiesis, it may indirectly control erythropoiesis by antagonizing Wnt/β-catenin signaling.45 Overall, Wnt/β-catenin signaling displays an evolutionary conserved molecular mechanism that dictates primitive hematopoiesis as seen in both in vivo and in vitro models of ES cell differentiation. Insights into the molecular mechanism that underlies Wnt/β-catenin-driven regulation of primitive hematopoiesis during embryogenesis are limited. A cross-talk between Wnt and BMP signaling by endoglin has been proposed to orchestrate primitive hematopoiesis.46-48 BMP and Wnt signaling cascades are 2 highly conserved signaling pathways that have been established to interact during many developmental processes, including regeneration of erythropoiesis upon BM irradiation.49 Endoglin, an ancillary receptor for several ligands of the transforming growth factor-β superfamily, is mostly known for its expression on vascular endothelium.50 Endoglin is critical for embryogenesis as endoglin-knockout mouse models fail to progress beyond 10.5 days of embryonic development, displaying vascular and cardiac abnormalities and anemia of the yolk sac.51 On the other hand, doxycycline-controlled upregulation of endoglin in ES cells enhances primitive hematopoietic differentiation and suppresses cardiac differentiation.46
Enforced expression of endoglin in ES cells hampers GSK3β activity that is dependent on Wnt/β-catenin signaling.46 Inhibition of GSK3β activity by canonical Wnt reflects a positive feedback signal to enforce Wnt/β-catenin signaling.52 The underlying mechanism by which endoglin is able to inhibit GSK3β activity, however, remains to be defined.46,48 Inhibition of GSK3β activity by endoglin accordingly activates BMP signaling through stabilization of Smad1.46 BMP signaling stimulates primitive hematopoiesis in cultured ES cells, in part by upregulation of Jun dimerization protein2 (JDP2) expression.46 JDP2 functions as either a transcriptional repressor or a coactivator, depending on cellular context, and thereby controls cell proliferation and differentiation.53 Since embryogenesis in JDP2-knockout transgenic mice is not affected, JDP2 appears to be dispensable for primitive hematopoiesis. Instead, JDP2 is revealed to control adipogenesis in these mice.54 Overall, spatiotemporal Wnt/β-catenin activity during embryonic development controls primitive hematopoiesis and promotes erythropoiesis during embryonic development. However, most of the underlying molecular mechanisms remain to be defined (Figure 1).
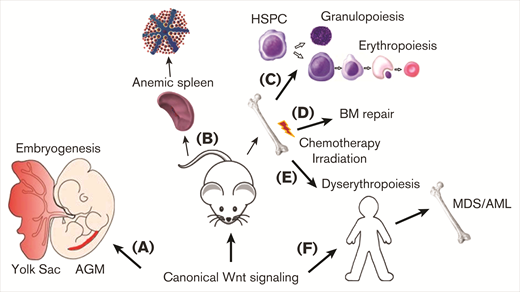
Wnt/β-catenin signaling controls erythropoiesis. Schematic overview reflecting the control of Wnt/β-catenin signaling on erythropoiesis. As reported during primitive hematopoiesis by stimulating primitive erythropoiesis in the yolk sac and by promoting erythroid progenitor formation during definitive hematopoiesis in the AGM region (A). Wnt/β-catenin also stimulates erythroid progenitor formation in the spleen of mice to promote stress erythropoiesis during anemia (B). Additionally, erythroid progenitor formation is supported by Wnt/β-catenin signaling by directing hematopoietic progenitor differentiation toward the erythroid lineage (C) and during BM injury to support recovery of erythropoiesis (D). Constitutive activation of Wnt/β-catenin in the hematopoietic lineage in mice however causes dyserythropoiesis (E), which correlates with MDS development and progression into AML (F).
Wnt/β-catenin signaling controls erythropoiesis. Schematic overview reflecting the control of Wnt/β-catenin signaling on erythropoiesis. As reported during primitive hematopoiesis by stimulating primitive erythropoiesis in the yolk sac and by promoting erythroid progenitor formation during definitive hematopoiesis in the AGM region (A). Wnt/β-catenin also stimulates erythroid progenitor formation in the spleen of mice to promote stress erythropoiesis during anemia (B). Additionally, erythroid progenitor formation is supported by Wnt/β-catenin signaling by directing hematopoietic progenitor differentiation toward the erythroid lineage (C) and during BM injury to support recovery of erythropoiesis (D). Constitutive activation of Wnt/β-catenin in the hematopoietic lineage in mice however causes dyserythropoiesis (E), which correlates with MDS development and progression into AML (F).
Canonical Wnt signaling controls erythropoiesis in response to anemia
Whereas primitive erythropoiesis is promoted by Wnt/β-catenin signaling, its importance during homeostatic definitive erythropoiesis in BM is less understood. Various studies investigated the control of Wnt/β-catenin signaling on HSC maintenance and differentiation in BM by conditional deletion of β-catenin or manipulation of Wnt regulators in transgenic mouse models.17,20,22,39,55 Hematopoietic-specific loss of β-catenin in transgenic Vav1-Cre;Ctnnb1F/F mice impairs HSC self-renewal, as observed using the Wnt-negative regulator Dickkopf 1 and Wnt3a-deficient mice.17,39,55 Conditional loss of β-catenin and combined loss with γ-catenin in HSCs in Mx1+-Cre;Ctnnb1F/F or Mx1+-Cre;Ctnnb1F/F;JupF/F transgenic mice, however, does not affect HSC behavior during steady-state hematopoiesis.23,24,56 These findings challenge the hypothesis that canonical Wnt signaling controls HSC maintenance.17,20,22,39,55 Gain-of-function approaches by introduction of stabilized forms of β-catenin exhaust the HSC pool, hamper reconstitution of the hematopoietic system upon transplantation, or enhance HSC function and maintenance by promotion of an immature phenotype.16,18,19,57 These contradictive findings may in part be explained by the different levels of signaling that are gained in vivo by the diverse strategies to induce canonical Wnt. Hypomorphic allele mutations and conditional deletion of one or both alleles of the adenomatous polyposis coli (APC) gene, a component of the β-catenin destruction complex, revealed mild to moderate activation of canonical Wnt and increases clonogenic and differentiation potential of HSCs.58 Enhanced Wnt/β-catenin activity on the on the other hand blocks HSC maintenance and fails to reconstitute irradiated recipient mice.19,58 These observations accordingly led to the “just-right” model that may explain the different cellular outcomes upon modulating canonical Wnt signaling.59 Keeping this model in mind, we will in the next part evaluate the control of canonical Wnt signaling on homeostatic erythropoiesis.
Whereas loss of β-catenin in embryonic endothelium inhibits HSC formation, thereby precluding initiation of erythropoiesis, canonical Wnt signaling is dispensable during definitive erythropoiesis in mouse BM.23,24 Inhibition of canonical Wnt signaling in HSCs by hematopoietic-specific loss of β-catenin in Vav1-Cre;Ctnnb1F/F transgenic mice or by transplanting Mx1+-Cre;Ctnnb1F/F-derived HSCs, does not alter erythroid lineage specification by HSCs or RBC formation.17,23 Additionally, combined loss of β-catenin with γ-catenin does not hamper erythropoiesis in mice either, indicating that erythropoiesis in steady-state conditions does not rely on β-catenin or redundant γ-catenin signaling.24
However, myeloablation by total body irradiation in mice triggers canonical Wnt signaling by widespread upregulation of Wnt10b ligand to promote expansion and erythroid lineage commitment of HSPCs.25 Canonical Wnt signaling indeed underlies BM recovery upon injury, as hematopoietic-specific depletion of β-catenin hampers hematopoietic recovery in mouse upon BM injury as induced by irradiation or chemotherapy.25 These findings are in line with the established role of canonical Wnt signaling to stimulate tissue repair as also seen during regeneration of bone mass.60-62 Wnt/β-catenin signaling directs cell fate decision of mesenchymal stem cells (MSCs) toward osteoblastogenesis at the cost of adipogenesis in order promote bone formation.60-62 Canonical Wnt signaling therefore seems dispensable for steady-state erythropoiesis but essential during recovery to steady-state erythropoiesis upon BM injury.
Besides BM repair, canonical Wnt signaling modulates extramedullary hematopoiesis in the anemic spleen in mice.26 Initiation of stress erythropoiesis in mice is induced in the spleen and characterized by a strong influx of monocytes that differentiate into nurse macrophages.63 The accumulation of nurse macrophages accordingly support the rapid expansion of erythroblastic islands that are required to generate an emergency wave of RBCs.63 Splenic macrophages secrete canonical Wnt ligands Wnt2b and Wnt8a to activate Wnt/β-catenin signaling in stress erythroid progenitors (SEPs) in order to promote SEP expansion.26 The molecular mechanism that underlies the production of canonical Wnt ligands in splenic monocytes and macrophages is unclear, but is likely controlled by hypoxic signaling as induced during anemia.64 Hypoxia-driven activation of canonical Wnt signaling is a highly conserved process described for various cell types ranging from healthy ES cells to tumor-initiating colon cells.65,66 Activation of canonical Wnt signaling is also followed by a hypoxic gene expression signature in EryP-CFCs in primitive erythropoiesis that develops under relative low-oxygen conditions in the yolk sac.28 Hypoxia can stimulate Wnt ligand production such as Wnt11 in various cell types and therefore may induce canonical Wnt ligand production in splenic monocytes and macrophages in the anemic spleen.67 Additionally, HIF1-α, a central downstream regulator of hypoxic signaling, is able to directly control Wnt signaling, as observed during development of the hippocampus using Cre-ERT2;Hif-1αF/F transgenic mice.68 Moreover, target genes of canonical Wnt signaling, such as TCF-1 and LEF-1, contain hypoxia response elements and thereby can be directly controlled by HIF1-α.6 5 HIF1-α therefore may control erythropoiesis by stimulating canonical Wnt signaling to drive erythroid progenitor expansion upon low oxygen tension as occurs in the spleen of anemic mice or in the yolk sac during the onset of primitive erythropoiesis. Canonical Wnt signaling during stress erythropoiesis in mice is indispensable, as loss of β-catenin in transplanted SEPs, derived from Cre-ERT2;Ctnnb1F/F transgenic mice, fail to provide erythroid short-term protection against radiation.26 The underlying mechanism by which canonical Wnt signaling promotes SEP expansion in anemic mice remains to be elucidated. Canonical Wnt signaling by itself seems insufficient to promote stress erythropoiesis in the hypoxic spleen, as SEPs also require BMP4, stem cell factor, and glucocorticoids to successfully expand in vitro.26,69,70
In mouse BM, constitutive activation of canonical Wnt signaling, by expression of a stable N-terminal deletion variant of β-catenin in Mx1-Cre;Ctnnb1(Ex3)Fl/Fl transgenic mice, severely hampers late-stage erythropoiesis.19 Constitutive active β-catenin signaling in these mice induces an accumulation of (pro)erythroblast and hampers their differentiation into reticulocytes.19 Similarly, a nondegradable active β-catenin S33Y mutant in mouse BM enforces the expansion of the (pro)erythroblasts.18 Aside expansion of (pro)erythroblasts, myeloid lineage formation in BM of these mice is severely hampered, and consequently, development of central macrophages is impaired.18,19 The interplay between the central macrophage and surrounding erythroid progenitors during erythropoiesis supports terminal differentiation of erythroblasts by phagocytosis of the pyrenocytes upon enucleation.71 Accumulation of (pro)erythroblasts upon enforced activation of canonical Wnt signaling may therefore partly relate to the reduced formation of central macrophages.18,19 Forced canonical Wnt signaling, however, also increases erythroid precursors formation in colony-formation assays for which expansion does not rely on the presence macrophages.18,19
Constitutive activation of Wnt/β-catenin signaling may shift HSPC commitment toward the erythroid cell lineage by altering expression of critical transcription factors that control cell fate decision.18 Gene expression analysis for example reveals Wnt/β-catenin signaling to downregulate Sfpi1, encoding PU.1, in HSPCs.18 PU.1 controls the activity of a wide variety of transcription factors that drive HSPC lineage commitment, such as GATA-1 that is essential to induce erythropoiesis.72 While PU.1 is known to block the transcriptional activity of GATA-1, reduced Sfpi1 expression may enhance erythroid lineage commitment of HSPCs. Yet, erythroblasts that develop from HSPCs by constitutively active β-catenin signaling reveal a subtle increase in PU.1 protein levels that at this stage may prevent terminal differentiation by inhibition of GATA-1.18
Besides deregulating transcription factors controlling hematopoietic cell fate decision, Wnt/β-catenin signaling may also stimulate (pro)erythroblast expansion by promoting cell-cycle progression. Wnt/β-catenin signaling promotes cell division by β-catenin-driven activation of G1 regulators such as cyclin-D1 and MYC that stimulate G1 progression toward S phase.4,73 Differentiation of CFU-Es toward (pro)erythroblasts is also driven during S phase.74 Wnt/β-catenin-driven G1 progression of early erythroid progenitors thereby may additionally promote the accumulation of (pro)erythroblasts as seen in BM of mice with constitutive activation of Wnt/β-catenin signaling.18,19
Finally, Wnt/β-catenin signaling may promote expansion of (pro)erythroblasts by supporting the high metabolic demand. Erythroblasts typically divide 3 or 4 times while differentiating toward reticulocytes.75 The high mitotic potential of erythroblasts demands large amounts of energy. Activation of Wnt/β-catenin signaling by expression of a constitutively active β-catenin variant in mouse liver cells strongly activates glutamine metabolism by inducing protein expression of glutamine synthesase, ornithine aminotransferase and glutamate transporter 1.76 Wnt/β-catenin signaling may similarly prevent energy shortage in erythroid precursors during anemia, but this remains to be investigated. Since glutamine constitutes the prime factor for hemoglobin synthesis, stimulation of glutamine metabolism by Wnt/β-catenin signaling may even enhance the production of hemoglobin to cope with the increased demand of RBCs during stress erythropoiesis.77 Overall, Wnt/β-catenin promotes accumulation of (pro)erythroblast in mouse BM. Constitutive activation of canonical Wnt, however, seems to hamper erythroblast differentiation and thereby may underlie dyserythropoiesis as seen in MDS patients (Figure 1).
Wnt/β-catenin signaling is deregulated in MDS
Deregulation of Wnt/β-catenin signaling has been implicated to support MDS development.8 MDS reflects a clonal stem cell disease displaying numerous genetic and epigenetic aberrations.78 Molecular mechanisms that underlie MDS formation are therefore still poorly understood. Among MDS patients, interstitial deletion on chromosome 5q (del5q) is the most common recurring cytogenetic abnormality as observed in 10% to 20% of primary MDS and 40% of therapy-related MDS/AML patients.79 Interstitial deletion in 5q affects the expression of several tumor suppressor genes in this region such as APC.6 Deficiency of APC results in β-catenin protein accumulation and activation Wnt/β-catenin signaling.58 Stabilization of endogenous β-catenin in APC haploinsufficient Mx1+-Cre;APC+/− transgenic mice hampers, similarly to constitutively stable β-catenin variants, erythroid differentiation at the CFU-E/(pro)-erythroblast stage in mouse BM, leading to severe anemia and death.5,18,19 Increased Wnt/β-catenin activity as a result of APC haploinsufficiency directly hampers the differentiation of CFU-E/(pro)erythroblasts, as heterozygous β-catenin deletion in APC haploinsufficient transgenic Apcdel/+, Ctnnb1del/+ mice restores erythropoiesis and prevents anemia.6 In contrast to enforced overexpression of active β-catenin variants in HSCs, APC haploinsufficient HSCs are still able to form monocytes and nurse macrophages.5 This difference can be explained by the just-right model indicating that a subtle activation of canonical Wnt signaling is sufficient to promote (pro)erythroblast expansion and decrease their differentiation, whereas monocyte formation is only impaired upon stronger Wnt/β-catenin activity. Overall, Wnt/β-catenin signaling controls erythropoiesis at the CFU-E/(pro)-erythroblast stage in mouse BM and thereby may underlie dyserythropoiesis in del5q MDS patients.
Asides genomic deletions, somatic genetic mutations in key components of the spliceosome machinery (eg, SF3B1, SRSF2, and U2AF1) and epigenetic regulators (eg, ASXL1, DMNT3A, and TET2) dominate the mutational landscape of MDS.80-82 Deregulation of spliceosome regulators by overexpression of SRSF4, SRSF6, or TRA2β promotes MYC-induced transformation of human MCF-10A mammary epithelial cells, which correlated with elevated Wnt/β-catenin signaling.82 Additionally, epigenetic deregulation of promoter methylation of Wnt inhibitors (eg, Wif-1, SFRP1, SFRR2, SFRP4, SFRP5, and DKK1) is frequently found in AML.81 This attenuates Wnt/β-catenin signaling and correlates with disease progression. Genetic mutations in components of the spliceosome and/or epigenetic regulators in MDS may therefore also promote Wnt/β-catenin activation in MDS.
Besides intracellular molecular alterations, paracrine aberrations have also been postulated to contribute to MDS formation.7,8,83-86 BM stroma of MDS patients reveals a deregulated canonical Wnt signature as defined by genome-wide sequencing of nonhematopoietic stromal cultures.8 Additionally, MSCs from MDS BM reveal promoter hypermethylation and underexpression of FRZB, a secreted Wnt antagonist.8 Increased canonical Wnt signaling in CD34+ HSCs due to reduced secretion of Wnt antagonists by the surrounding BM stroma therefore may also promote dyserythropoiesis in MDS patients.8 Dyserythropoiesis in MDS may also be driven by inflammation.87-89 Inflammatory cytokines frequently accumulate in BM of MDS patients and impair erythropoiesis.88,90-92 Tumor necrosis factor α, for example, blocks erythropoiesis by instructing HSPCs to differentiate toward the myeloid lineage, as induced by stimulation of PU.1 signaling.88
In addition, danger-associated molecular patterns (DAMPs), such as S100A8/A9, accumulate at high levels under inflammatory conditions in the plasma of MDS patients.90,91 DAMPs have been recognized to control hematopoiesis in part by regulating pyroptosis, an inflammatory-induced cell death program.87,93 DAMPs promote pyroptosis by stimulating the generation of reaction oxygen species, required to form cytosolic plaques that trigger Caspase-1-driven programmed cell death.87 Stimulation of pyroptosis by DAMPs may therefore also in part underlie dyserythropoiesis in MDS. Additionally however, treatment of healthy BM-derived mononuclear cells with S100A9 also promotes Wnt/β-catenin signaling.87 DAMP-driven reaction oxygen species activation stimulates stabilization and nuclear localization of β-catenin by dissociating nucleoredoxin from disheveled, that thereby inactivates the β-catenin destruction complex.94 Activation of Wnt/β-catenin signaling by inflammation may provide a repair response to regenerate BM that is damaged under inflammatory conditions, as seen during recovery of BM damage upon myeloablation or chemotherapy.25 Chronic inflammation, however, may lead to constitutive activation of Wnt/β-catenin signaling, which accordingly also may lead to dyserythropoiesis in MDS patients.
Gene expression analysis of CD34+ HSCs from more advanced cases of MDS reflect an active canonical Wnt signature, which is accordingly hypothesized to promote the oncogenic transformation of MDS toward AML.8 Conversion of MDS to AML is associated with MDMX overexpression, a p53 regulator.95 MDMX however also has been identified to sequester CK1α, a key component of the β-catenin destruction complex, that thereby prevents the degradation of β-catenin.14 The notion that MDMX is overexpressed in the vast majority of AML patients (>90%), identified MDMX overexpression to accelerate AML formation in preleukemic Flt3WT/ITD, URE−/−, and Tet2−/− transgenic mouse models that are used to model MDS/AML formation.14 MDMX overexpression in HSCs of transgenic mouse models and leukemic cell lines accordingly increases protein accumulation and nuclear translocation of β-catenin, thereby altering gene expression, independent of p53.14 HSCs obtained from high-risk MDS patients, displaying a high potential to develop AML, are also marked by increased MDMX expression and an active canonical Wnt gene expression signature.14 Overall, this indicates that unrestrained activation of Wnt/β-catenin signaling may underlie dyserythropoiesis in MDS and promote the transformation of MDS to AML (Figure 2). These findings raise the question whether current treatments for MDS affect Wnt/β-catenin signaling. Luspatercept has been found to induce transfusion independency for at least 8 weeks in ∼40% of low-risk MDS patients compared with 13% of patients that were treated with placebo.96 Luspatercept resembles an activin receptor fusion protein that promotes erythropoiesis by trapping BMP/transforming growth factor β (TGF-β) ligands.96,97 BMP/TGF-β and Wnt/β-catenin signaling closely cooperate to balance cell proliferation and differentiation as outlined above.46,49 Inhibition of BMP/TGF-β signaling by luspatercept may therefore partially dampen Wnt/β-catenin signaling in del5q low-risk MDS patients.49 Azacitidine is currently standard treatment of high-risk MDS, despite the fact that many patients do not respond to this treatment.98 Azacitidine represents a hypomethylating agent that may inhibit Wnt/β-catenin activity by demethylating promoter elements of Wnt inhibitors (eg, DKK1), as revealed in the MDS-derived SKM-1 cell line.99 Overall, the clinical response to current MDS treatments may correlate with inhibition of Wnt/β-catenin signaling and can possibly be used to predict clinical outcome.
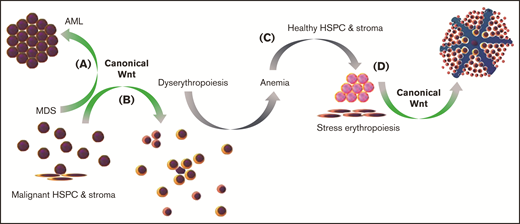
Deregulation of Wnt/β-catenin signaling in MDS. Schematic overview proposing chronic activation of canonical Wnt/β-catenin signaling to promote transformation of MDS to AML (A) and underlie dyserythropoiesis in MDS (B), leading to anemia (C) and triggering stress erythropoiesis by healthy HSPCs and stroma that remain present in the BM (D), which is also promoted by canonical Wnt/β-catenin signaling. Manipulation of canonical Wnt/β-catenin signaling in MDS may therefore, in parallel, alter stress erythropoiesis and increase anemia severity.
Deregulation of Wnt/β-catenin signaling in MDS. Schematic overview proposing chronic activation of canonical Wnt/β-catenin signaling to promote transformation of MDS to AML (A) and underlie dyserythropoiesis in MDS (B), leading to anemia (C) and triggering stress erythropoiesis by healthy HSPCs and stroma that remain present in the BM (D), which is also promoted by canonical Wnt/β-catenin signaling. Manipulation of canonical Wnt/β-catenin signaling in MDS may therefore, in parallel, alter stress erythropoiesis and increase anemia severity.
Concluding remarks
In summary, spatiotemporal activation of canonical Wnt promotes erythroid progenitor formation during primitive hematopoiesis and underlies the recovery of erythropoiesis upon BM damage.18,19,25,28,32 Chronic activation of canonical Wnt deregulates erythropoiesis and thereby may cause dyserythropoiesis in MDS patients.5-7,10 Moreover, prolonged Wnt/β-catenin activation seems to promote transformation of MDS to AML.8,14 Wnt/β-catenin signaling accordingly may display a therapeutic target to treat MDS and prevent leukemia formation. However, MDS is characterized by anemia, which triggers stress erythropoiesis by healthy HSPCs that are still present.15 These healthy HSPCs may require β-catenin signaling in order to establish an emergency response (Figure 2). Manipulation of β-catenin signaling in MDS therefore could deregulate stress erythropoiesis and consequently increase anemia severity. Yet, the control of canonical Wnt on stress erythropoiesis has only been reported in mice.26 In mice, stress erythropoiesis primarily develops in the spleen, in contrast to the BM, as reported for humans.100 This difference therefore questions how canonical Wnt signaling underlies stress erythropoiesis in human BM. Our laboratory and others revealed E-cadherin to distinguish human from mouse erythroid progenitors.101,102 In human BM, E-cadherin is selectively expressed in the erythroid lineage which is not conserved in mouse (R.E.K., Han Verhagen, Melli Xia, Marja Nieuwland, Iris Rink, Ron Kerkhoven, Carlijn Voermans, Emile van den Akker, Marieke von Lindern and M.N., manuscript in preparation).101,102 E-cadherin interacts with canonical Wnt signaling by competing with APC for binding β-catenin, thereby controlling β-catenin signaling.103-107 In epithelia, increased tension on the cell surface enhances E-cadherin clustering, thereby driving cell proliferation through downstream activation of β-catenin signaling.104,106,107 Likewise, activation of β-catenin by mechanical strain in BM is shown to promote osteoblast formation.60-62 Mechanical induction of β-catenin can also act in synergy with canonical Wnt signaling to stimulate cell proliferation of epithelial cells or instruct differentiation of MSCs.104,106,107 E-cadherin expression by human erythroblasts suggests an interplay between mechanotransduction and canonical Wnt signaling to control erythropoiesis in human BM.
In conclusion, whereas β-catenin signaling may display a therapeutic candidate to treat MDS, there is an urgent need to improve our understanding of the contribution and underlying molecular mechanism of Wnt/β-catenin signaling to (stress) erythropoiesis in human BM in order to develop novel opportunities for MDS treatment.
Acknowledgments
The authors thank Emile van den Akker (Sanquin Research and Landsteiner Laboratory, Amsterdam UMC, The Netherlands) for the discussion on primitive hematopoiesis and Arjan van de Loosdrecht (Amsterdam UMC, The Netherlands) for providing discussion on MDS. Finally, the authors apologize that they cannot cite all the excellent literature that is connected to the topic of this review due to space limitations.
Research in the Nethe laboratory is funded by Joghem van Loghem fellowship grant #00113.
Authorship
Contribution: M.N. prepared the original draft; and R.A.K. and M.N. reviewed and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing interests.
Correspondence: Micha Nethe, Department of Hematopoiesis, Sanquin Research and Landsteiner Laboratory, Amsterdam UMC, Plesmanlaan 125, 1181 SL Amsterdam, The Netherlands; e-mail: m.nethe@sanquin.nl.

