

Key Points
Membrane SM reduction by SMS1deficiency enhances PS exposure and thrombocytopenia.
Depression of membrane SM potentiates Ca2+ influx and PS externalization through TMEM16F.

Abstract
Sphingomyelin synthase 1 (SMS1) contributes to the generation of membrane sphingomyelin (SM) and affects SM-mediated physiological functions. Here, we describe the hematologic phenotypes, such as reduced circulating platelets and dysfunctional hemostasis, in SMS1-deficient (SMS1-KO) mice. SMS1-KO mice display pathologic manifestations related to idiopathic thrombocytopenia (ITP), including relatively high amounts of peripheral blood reticulated platelets, enhanced megakaryopoiesis in the bone marrow and spleen, and splenomegaly. Deficiency of SMS1, but not SMS2, prevented SM production and enhanced phosphatidylserine (PS) externalization on the plasma membranes of platelets and megakaryocytes. Consequently, SMS1-KO platelets were excessively cleared by macrophages in the spleen. Multimer formation in the plasma membrane of TMEM16F, a known calcium (Ca2+)-activated nonselective ion channel and Ca2+-dependent PS scramblase, was enhanced; the result was PS externalization to outer leaflets through increased Ca2+ influx in immortalized mouse embryonic fibroblasts established from SMS1-KO mice (SMS1-KO tMEFs), as seen with SMS1-KO platelets. Thus, SMS1 deficiency changed the TMEM16F distribution on the membrane microdomain, regulating Ca2+ influx-dependent PS exposure. SMS1-KO tMEFs in which TMEM16F was knocked out by using the CRISPR/Cas9 system lacked both the Ca2+ influx and excess PS exposure seen in SMS1-KO tMEFs. Therefore, SM depletion on platelet membrane microdomains due to SMS1 deficiency enhanced PS externalization via a Ca2+ influx through TMEM16F activation, leading to elevated platelet clearance and causing hemostasis dysfunction through thrombocytopenia. Our current findings show that the SM-rich microdomain generated by SMS1 is a potent regulator of thrombocytopenia through TMEM16F, suggesting that its dysfunction may be a novel additional mechanism of ITP.
Introduction
Membrane lipids such as phosphatidylserine (PS) localize asymmetrically, and PS dynamism between the plasma membrane inner and outer leaflets is implicated in various cellular functions. PS mainly localizes in the plasma membrane inner leaflets and is exposed on outer leaflets in response to extracellular stimuli, inducing apoptosis.1 In apoptotic cells, exposed PS acts as an “eat-me” signal to induce phagocytosis by macrophages.2 Similarly, PS exposure on plasma membrane outer leaflets in platelets plays important roles in coagulation and clearance.3,4 Continuous PS externalization causes platelet apoptosis and subsequent thrombocytopenia due to elevated platelet clearance in the spleen. For example, inhibition of the antiapoptotic Bcl-2 family protein Bcl-xL, which normally inhibits apoptotic proteins such as Bak or Bax, leads to reduced platelet life spans and thrombocytopenia with increased PS exposure.5,6 Inversely, the deletion of both Bak and Bax elongates platelet half-life and prevents the thrombocytopenia induced by Bcl-xL inhibition.7,8 Therefore, regulation of membrane PS externalization in the apoptosis process determines platelet life span by accelerating platelet clearance.
In platelets, PS exposure is mainly regulated by TMEM16F and calcium (Ca2+) influx.9 TMEM16F was first found as a Ca2+-activated cation channel and recently recognized as a Ca2+-activated phospholipid scramblase.10,11 Heterozygous TMEM16F mutations were found in patients with Scott syndrome, in whom the defective TMEM16F function induced a bleeding disorder due to defective activation of coagulation factors Va and Xa via impaired PS exposure on platelets.9,12 Inversely, TMEM16F overexpression in Ba/F3 cells robustly induced PS externalization.9
In contrast with PS, sphingomyelin (SM) mainly localizes in plasma membrane outer leaflets and forms a major microdomain (lipid raft) compartment defined as a detergent-resistant membrane (DRM).13 SM-rich DRMs play pivotal roles in diverse cellular functions such as cell proliferation, migration, and inflammation.14-16 Membrane SM levels affect the PS homeostasis levels in cells. Gulshan et al17 reported that spontaneous translocation of external PS into the inner-membrane by flippase was regulated by SM in a cell-free system. In addition, membrane SM depletion increased extracellular PS levels by inhibiting flipping in HEK293 cells expressing the Tangier disease W590S mutant ABCA1 isoform. However, the pathophysiological roles of SM and SM-rich DRM-dependent PS externalization in regulating platelet coagulation and clearance are largely unknown.
SM is synthesized from ceramide and phosphatidylcholine by SM synthases (SMSs), which have 2 isoforms, SMS1 and SMS2.18 SMS1 localizes to the Golgi apparatus, and SMS2 is distributed in both the Golgi apparatus and the plasma membrane. Previous investigations have provided evidence that SM synthesized by both SMS1 and SMS2 in the Golgi apparatus is transported to cellular membranes, including the plasma membrane, whereas SMS2-generated SM on the plasma membrane is regulated in response to extracellular stimuli.18 To elucidate the physiological roles of SM and SMSs, mice deficient in Sgms1 (SMS1-KO) or Sgms2 (SMS2-KO) have been established, and their phenotypes examined. Both SMS1-KO and SMS2-KO mice have reduced inflammatory responses and ameliorated inflammatory diseases, including atherosclerosis, hepatitis, and type 2 diabetes, owing to inflammatory chemokine/cytokine suppression resulting from decreased membrane SM.19-22 Similarly, our previous studies using SMS-KO mice showed that membrane SM generated by SMS1 and SMS2 regulated pathophysiological functions in diverse disease models: (1) because Japanese encephalitis virus (JEV) infects target cells by attaching through membrane SM, JEV infection was inhibited in SMS1-KO mice23 ; and (2) SMS2-dependent membrane SM regulated the transcriptional expression of cell–cell adhesion molecule ICAM-1, leading to enhanced lymphoma cell infiltration into the mouse liver.24
Regarding blood coagulation, SMS1-generated SM-rich DRMs supply the location for promoting clot retraction via the enhancement of fibrin-αIIbβ3-myosin complex formation, regardless of PS externalization.25 Consequently, SMS1-deficient extracted platelets exhibit delayed clot retraction. However, the pathophysiological relevance of SMS deficiency for the platelet life cycle, including thrombocytosis and megakaryocyte (MK) production and clearance, is completely unclear. Immune (idiopathic) thrombocytopenia (ITP) has been recognized as a complex autoimmune disease characterized by decreased peripheral blood platelet numbers.26,27 The causes of primary ITP remain unclear, but antiplatelet autoantibody generation by plasmacytes, T cell–mediated platelet destruction, and impaired MK function are believed to be key mechanisms. In contrast, secondary ITP is due mainly to chronic infections, such as by Helicobacter pylori, and autoimmune diseases, such as systemic lupus erythematosus or rheumatoid arthritis. The first choice of medication for ITP is corticosteroids, but some cases exhibit steroid-resistant thrombocytopenia, the pathology of which has not been clarified. Here, we suggest that PS externalization through TMEM16F resulting from an SMS1-dependent SM deficiency in the plasma membrane microdomain is a possible cause of ITP unrelated to either immune impairment or chronic infection.
Materials and methods
Mice
The generation of mice lacking Sgms1 (SMS1-KO) and Sgms2 (SMS2-KO) was conducted as previously described.23,24 The C57BL/6 wild-type (WT) mice used here were littermates of SMS1-KO mice or purchased from Clea Japan (Tokyo, Japan). Adult mice (aged >12 weeks) were used for all studies. All experiments were approved by the Committee for Animal Experiments of Kanazawa Medical University and were performed in accordance with the Guidelines for Animal Experimentations of Kanazawa Medical University.
The splenectomy procedure was conducted as described elsewhere; details are available in the supplemental Materials and methods.28
Hematologic analysis and platelet/MK isolation
These protocols are available in the supplemental Materials and methods.
Histology, immunohistochemistry, and immunocytochemistry
These protocols are available in the supplemental Materials and methods.
Flow cytometry
The measurements of membrane SM and PS were conducted as previously described.23,29 Protocols are available in the supplemental Materials and methods.
Liquid chromatography-tandem mass spectrometry
To detect lipids, including SM and ceramide, liquid chromatography–electrospray ionization mass spectrometry (Ultimate 3000 LC system, Thermo Fisher Scientific, Waltham, MA) was performed as previously described.30
Platelet life span assay
The analysis of platelet life span was conducted as described previously.31,32 The protocol is available in the supplemental Materials and methods.
Cell culture and calcium imaging
Mouse embryonic fibroblasts immortalized by SV40 large T antigen (tMEFs) were previously established from WT, SMS1-KO, and SMS2-KO mice.16 Protocols are available in the supplemental Materials and methods.
DRM fractionation and western blot analysis
Extraction of the DRM fraction was performed as previously described.16 Protocols are available in the supplemental Materials and methods.
Statistical analysis
Statistical analysis was performed by using StatView 5.0 software (SAS Institute, Inc., Cary, NC). The results are presented as the mean ± standard deviation. Data were assessed by using a Student t test or analysis of variance. P values <.05 were considered to indicate statistically significant differences.
Results
SMS1-deficient mice manifest thrombocytopenia
To investigate the effects of SMS on hemocyte characteristics, we performed a complete blood count on peripheral blood collected from mice. SMS1-KO mice exhibited lower platelet numbers (35.08 ± 4.65 × 104 cells/µL) compared with both WT (93.42 ± 25.06 × 104 cells/µL) and SMS2-KO (93.42 ± 25.06 × 104 cells/µL) mice (Figure 1A), whereas the numbers of red and white blood cells did not differ (Figure 1B-C). To examine the effect on hemostasis of decreased platelets in SMS1-KO mice, a tail vein bleeding time assay was performed. Although tail vein bleeding was stopped within 150 seconds in WT and SMS2-KO mice, the SMS1-KO mice exhibited a prolonged bleeding time of >300 seconds (Figure 1D). Previously, we established mice with a conditional Sgms1 allele (SMS1fl/fl) and crossed SMS1fl/fl mice with Rosa-CreER transgenic mice, which express CreER recombinase and allow the deletion of Sgms1 allele by tamoxifen treatment (Rosa-CreER;SMS1fl/fl) (supplemental Figure 1A).33 After a tamoxifen injection, Rosa-CreER;SMS1fl/fl mice but not SMS1fl/fl mice exhibited Sgms1 allele deletion (supplemental Figure 1B). Tamoxifen-treated Rosa-CreER;SMS1fl/fl mice had lower platelet numbers compared with not only control mice (without tamoxifen treatment) but also with tamoxifen-treated SMS1fl/fl mice; however, their red and white blood cell counts were not different (supplemental Figure 1C).
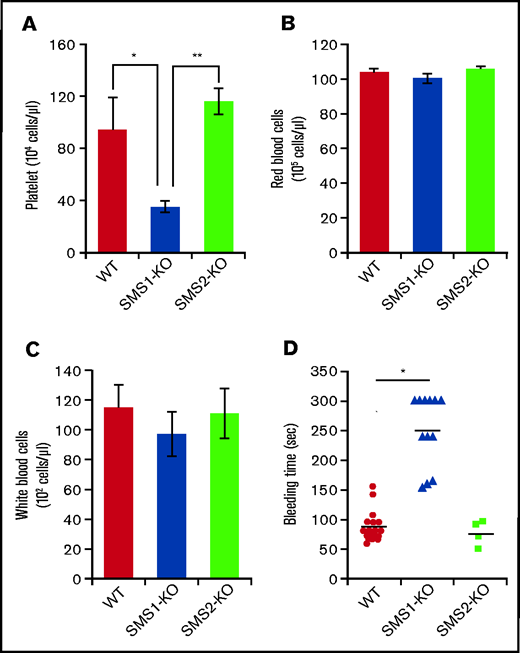
Platelet numbers and bleeding time in WT, SMS1-KO, and SMS2-KO mice. (A-C) Complete blood count analyses were performed on whole blood extracted from WT, SMS1-KO, and SMS2-KO mice (n = 3 per group). The numbers of platelets (A), red blood cells (B), and white blood cells (C) in mice of each genotype are shown. (D) The tail vein bleeding times of WT (n = 18), SMS1-KO (n = 12), and SMS2-KO (n = 4) mice are shown. Values show the mean ± standard deviation. *P < .05; **P < .005.
Platelet numbers and bleeding time in WT, SMS1-KO, and SMS2-KO mice. (A-C) Complete blood count analyses were performed on whole blood extracted from WT, SMS1-KO, and SMS2-KO mice (n = 3 per group). The numbers of platelets (A), red blood cells (B), and white blood cells (C) in mice of each genotype are shown. (D) The tail vein bleeding times of WT (n = 18), SMS1-KO (n = 12), and SMS2-KO (n = 4) mice are shown. Values show the mean ± standard deviation. *P < .05; **P < .005.
Reticulated platelets are the youngest circulating platelets, and their elevation reflects an enhancement of platelet production in thrombocytopenia.34,35 Determination of reticulated platelets stained by thiazole orange showed that SMS1-KO mice had higher amounts of reticulated platelets in their peripheral blood (Figure 2A). Bone marrow is the main location for thrombopoiesis and megakaryopoiesis. However, thrombocytopenia enhances megakaryopoiesis to recruit fewer platelets from MKs in not only the bone marrow but also ectopically in the spleen.36 To examine whether SMS1 deficiency enhances megakaryopoiesis based on the increased production of reticulated platelets, we histologically observed spleen and tibial bone marrow staining by von Willebrand factor (vWF), which is a reliable MK immunohistochemical marker.37 Splenic MKs were detectable in SMS1-KO mice, whereas vWF-positive MKs were not observed in WT or SMS2-KO mice (Figure 2B). Moreover, the number of MKs in the tibial bone marrow was also higher in SMS1-KO mice compared with WT and SMS2-KO mice (Figure 2C). In some cases, splenomegaly appeared along with thrombocytopenia, owing to abnormal megakaryopoiesis and elevated platelet clearance.6,38 As expected, only SMS1-KO mice exhibited splenomegaly; WT and SMS2-KO mice did not (Figure 2D). These data suggest that SMS1 deficiency causes the obvious phenotype of thrombocytopenia, accompanied with an elevated platelet-producing ability.
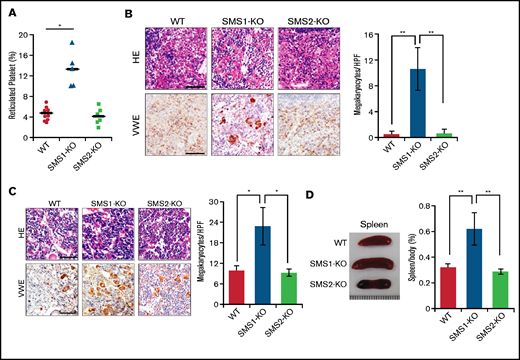
Assessment of extramedullary hematopoiesis and splenomegaly. (A) The reticulated platelets of WT (n = 10), SMS1-KO (n = 5), and SMS2-KO (n = 6) mice were stained with thiazole orange and analyzed by using flow cytometry. Paraffin sections of spleen (B) and bone marrow (C) were stained with hematoxylin and eosin (HE) or with anti-vWF antibody. The vWF-positive MKs in several randomly chosen high-power fields (HPF) were quantified. Scale bars, 50 µm. (D) Representative images of spleens from WT, SMS1-KO, and SMS2-KO mice. The ratios of the spleen to whole body weight were calculated in nine WT, ten SMS1-KO, and seven SMS2-KO mice. Values show the mean ± standard deviation. *P < .05; **P < .005.
Assessment of extramedullary hematopoiesis and splenomegaly. (A) The reticulated platelets of WT (n = 10), SMS1-KO (n = 5), and SMS2-KO (n = 6) mice were stained with thiazole orange and analyzed by using flow cytometry. Paraffin sections of spleen (B) and bone marrow (C) were stained with hematoxylin and eosin (HE) or with anti-vWF antibody. The vWF-positive MKs in several randomly chosen high-power fields (HPF) were quantified. Scale bars, 50 µm. (D) Representative images of spleens from WT, SMS1-KO, and SMS2-KO mice. The ratios of the spleen to whole body weight were calculated in nine WT, ten SMS1-KO, and seven SMS2-KO mice. Values show the mean ± standard deviation. *P < .05; **P < .005.
SMS1 massively contributes to the maintenance of membrane SM in platelets and MKs
SMS1 is responsible for the bulk of SM production to regulate numerous cellular functions.18,25 To examine the SMS1 contribution to generating plasma membrane SM, we detected membrane SM using lysenin-conjugated Venus (Venus-lysenin), an SM-specific probe, in CD41+ platelets (Figure 3A). Cell surface SM on platelets was minimal in SMS1-KO mice compared with WT mice (Figure 3B). However, SMS2 deficiency had no effect on the suppression of membrane SM production. To clarify the SM reduction in SMS1-KO mouse platelets, the amounts of lipids, including SM, were measured by using liquid chromatography–tandem mass spectrometry. Platelet SM was lower in SMS1-KO mice compared with WT and SMS2-KO mice (Figure 3C), whereas ceramide, which is the substrate of SM production, was not different among these mice (supplemental Figure 2A). To confirm the SM reduction in MKs, we isolated MKs from mouse tibias and femurs, then stained them with anti-CD41 antibody and Venus-lysenin. As with the platelets, bone marrow MKs from SMS1-KO mice had lower membrane SM levels compared with those from WT mice or SMS2-KO mice (Figure 3D). These data suggest that SMS1 contributes to membrane SM generation in both platelets and MKs.
![Generation of membrane SM in platelets and MKs. (A) Platelets were detected by using phycoerythrin-conjugated anti-CD41 antibody (anti–CD41-phosphatidylethanolamine [PE]) and flow cytometry. (B) Cell surface SM (stained with Venus-lysenin) in CD41+platelets from WT (n = 3), SMS1-KO (n = 4), or SMS2-KO (n = 4) mice was analyzed by flow cytometry and quantified as the mean fluorescent intensity (MFI). NS, no staining. (C) SM amounts in platelets from WT (n = 4), SMS1-KO (n = 6), or SMS2-KO (n = 5) mice were measured by using liquid chromatography–tandem mass spectrometry. (D) MKs were extracted from bone marrow cells, then stained with Venus-lysenin and anti–CD41-PE. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI), and images were obtained with confocal microscopy. The fluorescence intensity of Venus-lysenin was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD) and is indicated as arbitrary units (AU). Bars, 20 µm. DIC, Differential interference contrast. Values show the mean ± standard deviation. *P < .05; **P < .005.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/20/10.1182_bloodadvances.2020002922/4/m_advancesadv2020002922f3.png?Expires=1769134053&Signature=cx1kjRCmWzi~W8vWVgnnAkoPEIxi7uPxub6SXmD8sjJgRMZa6MwjQhDBNMrlkbdMZlAceiy0s28jnx0U5KPJhPGgcYb9s7kFei~GyFKVmcO1E9csQ093K7p9q3SqaOftiZ9HhDtucxsqWFrIfa5NsokVVbcF8rLEcjBQKWa9aYuKqDyCG9D7uIeFtLaODKFf3J7xtzWt1WnTw34-gH-UvUfh6jQnW93GVSKm-eaXnB-jGmNFpQM9DacliErOJxTC2ShuOkqIahm1rWKlDdKlQnQMgd~UBKPDp-O6UmPLQ8h8JLcBGLBi1g-IraJsoCFaNe9vW0KtTSBvG7wpKdL2Vg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Generation of membrane SM in platelets and MKs. (A) Platelets were detected by using phycoerythrin-conjugated anti-CD41 antibody (anti–CD41-phosphatidylethanolamine [PE]) and flow cytometry. (B) Cell surface SM (stained with Venus-lysenin) in CD41+platelets from WT (n = 3), SMS1-KO (n = 4), or SMS2-KO (n = 4) mice was analyzed by flow cytometry and quantified as the mean fluorescent intensity (MFI). NS, no staining. (C) SM amounts in platelets from WT (n = 4), SMS1-KO (n = 6), or SMS2-KO (n = 5) mice were measured by using liquid chromatography–tandem mass spectrometry. (D) MKs were extracted from bone marrow cells, then stained with Venus-lysenin and anti–CD41-PE. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI), and images were obtained with confocal microscopy. The fluorescence intensity of Venus-lysenin was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD) and is indicated as arbitrary units (AU). Bars, 20 µm. DIC, Differential interference contrast. Values show the mean ± standard deviation. *P < .05; **P < .005.
Generation of membrane SM in platelets and MKs. (A) Platelets were detected by using phycoerythrin-conjugated anti-CD41 antibody (anti–CD41-phosphatidylethanolamine [PE]) and flow cytometry. (B) Cell surface SM (stained with Venus-lysenin) in CD41+platelets from WT (n = 3), SMS1-KO (n = 4), or SMS2-KO (n = 4) mice was analyzed by flow cytometry and quantified as the mean fluorescent intensity (MFI). NS, no staining. (C) SM amounts in platelets from WT (n = 4), SMS1-KO (n = 6), or SMS2-KO (n = 5) mice were measured by using liquid chromatography–tandem mass spectrometry. (D) MKs were extracted from bone marrow cells, then stained with Venus-lysenin and anti–CD41-PE. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI), and images were obtained with confocal microscopy. The fluorescence intensity of Venus-lysenin was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD) and is indicated as arbitrary units (AU). Bars, 20 µm. DIC, Differential interference contrast. Values show the mean ± standard deviation. *P < .05; **P < .005.
Enhanced PS exposure on both platelets and MKs from SMS1-KO mice
Previous reports have shown that SM depletion impairs the PS inward translocation from plasma membrane outer leaflets to inner leaflets,17 and the increased PS externalization on circulating platelets promotes clearance by phagocytosis, leading to thrombocytopenia.39 We therefore hypothesized that the SM reduction resulting from SMS1 deficiency would enhance PS exposure on platelet membranes. To investigate this theory, we examined cell surface PS on platelets via staining with annexin V, a PS-specific probe. In WT and SMS2-KO mice, the levels of annexin V–positive platelets were low (11.33 ± 1.23% and 15.71 ± 2.83%, respectively) (Figure 4A). In contrast, SMS1-KO mice exhibited higher numbers of PS-externalized platelets (41.13 ± 2.56%), despite showing minimal or no change in the PS externalization of their splenic lymphocytes and peripheral erythrocytes (supplemental Figure 3). As on the platelets, PS exposure on the plasma membrane outer leaflet of bone marrow–derived MKs was also higher for SMS1-KO mice compared with WT and SMS2-KO mice (Figure 4B). These results suggest that SMS1 deficiency changes the polarity of PS on the plasma membrane and makes it easier for PS to localize to the outer membrane via SM reduction.
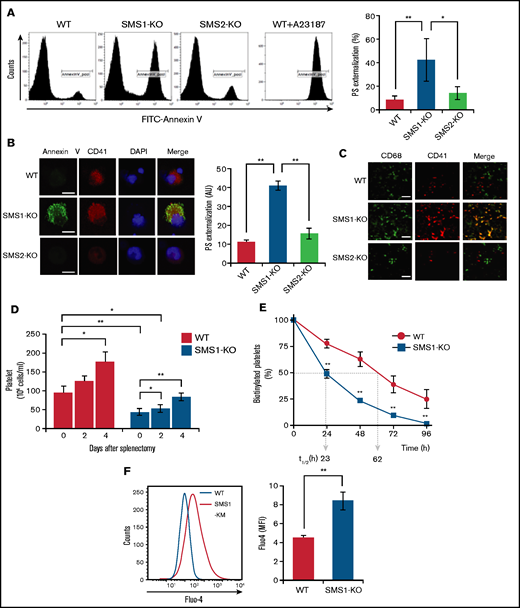
PS exposure and Ca2+ influx in platelets and MKs. (A) Membrane PS on platelets from SMS1-KO or WT mice (n = 3 per group) was stained with FITC-conjugated annexin V (FITC–annexin V) and detected by using flow cytometry. WT mouse platelets were pretreated with calcium ionophore A23187 for 10 minutes before PS staining. The percentages of FITC-annexin V–positive platelets (CD41+) are presented. (B) MKs were stained with FITC–annexin V and anti–CD41-phosphatidylethanolamine (PE). The nuclei were then counterstained with 4,6-diamidino-2-phenylindole (DAPI), and images were obtained with confocal microscopy. The fluorescent intensity of FITC–annexin V was quantified with ImageJ software and is presented in arbitrary units (AU). Scale bar, 20 µm. Values show the mean ± standard deviation. (C) Sections of mouse spleen were stained with Alexa Fluor 488–conjugated anti-CD68 antibody and anti–CD41-PE, and then observed with fluorescent microscopy. Scale bar, 50 µm. (D) Splenectomy was performed in WT and SMS1-KO mice (n = 3-5 each). Blood was then collected from the tail vein, and platelet numbers were counted on the indicated day after surgery. Values show the mean ± standard deviation. (E) WT (n = 5) and SMS1-KO (n = 3) mice were intravenously injected with N-hydroxysuccinimide ester (NHS)-conjugated biotin. Platelets were then isolated from whole blood collected at the indicated time points and stained with FITC-conjugated anti-CD41 antibody and allophycocyanin-conjugated streptavidin. The biotinylated platelets were analyzed with flow cytometry. t1/2 represents the half-life of platelets in hours. Values show the mean ± standard deviation. (F) Platelets from WT (n = 4) and SMS1-KO (n = 4) mice were stained with Furo-4 AM and anti–CD41-PE and then analyzed with flow cytometry. The intracellular Ca2+ levels in platelets were detected by measuring the Furo-4 AM fluorescence intensity and are presented as mean fluorescent intensity (MFI). Values show the mean ± standard deviation. *P < .05; **P < .005.
PS exposure and Ca2+ influx in platelets and MKs. (A) Membrane PS on platelets from SMS1-KO or WT mice (n = 3 per group) was stained with FITC-conjugated annexin V (FITC–annexin V) and detected by using flow cytometry. WT mouse platelets were pretreated with calcium ionophore A23187 for 10 minutes before PS staining. The percentages of FITC-annexin V–positive platelets (CD41+) are presented. (B) MKs were stained with FITC–annexin V and anti–CD41-phosphatidylethanolamine (PE). The nuclei were then counterstained with 4,6-diamidino-2-phenylindole (DAPI), and images were obtained with confocal microscopy. The fluorescent intensity of FITC–annexin V was quantified with ImageJ software and is presented in arbitrary units (AU). Scale bar, 20 µm. Values show the mean ± standard deviation. (C) Sections of mouse spleen were stained with Alexa Fluor 488–conjugated anti-CD68 antibody and anti–CD41-PE, and then observed with fluorescent microscopy. Scale bar, 50 µm. (D) Splenectomy was performed in WT and SMS1-KO mice (n = 3-5 each). Blood was then collected from the tail vein, and platelet numbers were counted on the indicated day after surgery. Values show the mean ± standard deviation. (E) WT (n = 5) and SMS1-KO (n = 3) mice were intravenously injected with N-hydroxysuccinimide ester (NHS)-conjugated biotin. Platelets were then isolated from whole blood collected at the indicated time points and stained with FITC-conjugated anti-CD41 antibody and allophycocyanin-conjugated streptavidin. The biotinylated platelets were analyzed with flow cytometry. t1/2 represents the half-life of platelets in hours. Values show the mean ± standard deviation. (F) Platelets from WT (n = 4) and SMS1-KO (n = 4) mice were stained with Furo-4 AM and anti–CD41-PE and then analyzed with flow cytometry. The intracellular Ca2+ levels in platelets were detected by measuring the Furo-4 AM fluorescence intensity and are presented as mean fluorescent intensity (MFI). Values show the mean ± standard deviation. *P < .05; **P < .005.
Enhanced platelet and MK clearance by macrophages in SMS1-KO mouse spleens
The appearance of PS on cell surfaces serves as a well-known signal for phagocytosis and the subsequent clearance of apoptotic cells.40 In addition, PS exposure on the platelet membrane also acts as a “clear-me” sign, and PS-exposing platelets are similarly eliminated by phagocytosis.4,40 Our data further exhibit enhanced PS externalization on both platelets and MKs from SMS1-KO mice (Figure 4A-B). Therefore, to determine if platelet clearance in SMS1-KO mice occurs because of increased phagocytosis, we observed the localization of CD68+macrophages and CD41+platelets/MKs in mouse spleens by immunohistochemistry. In WT and SMS2-KO mouse spleens, the numbers of both macrophages and platelets/MKs were low, and minimal colocalization between them was observed (Figure 4C). Notably, SMS1 deficiency not only increased the numbers of these cells, it also resulted in their colocalization in the spleen. These data suggest that enhanced platelet clearance in the spleen contributes to the thrombocytopenia of SMS1-KO mice.
To further investigate this mechanism, we performed splenectomies, which are clinically used to inhibit the clearance of platelets or red blood cells,41 with the aim of eliminating platelet and MK degradation by phagocytosis. In both WT and SMS1-KO mice, the number of circulating platelets appeared to increase at 4 days’ post-splenectomy compared with that pre-splenectomy (on day 0) (Figure 4D; supplemental Figure 4A). In addition, the numbers of red blood cells were increased in both WT and SMS1-KO mice from 2 days’ post-splenectomy (supplemental Figure 4B). Interestingly, post-splenectomy (day 4), SMS1-KO mice recovered their platelet numbers up to those of control WT mice (day 0).
We also investigated the platelet life-span by tracing biotin-labeled platelets with streptavidin staining.31,32 The half-life (t1/2; clearance of biotin-labeled platelets) of platelets from WT mice (62 hours) (Figure 4E; supplemental Figure 5) was longer than that of platelets from SMS1-KO mice (23 hours). Together, these data suggest that thrombocytopenia in SMS1-KO mice is at least partially caused by an accelerated clearance of platelets/MKs via phagocytosis conducted by macrophages in the spleen.
SM depletion due to SMS1 deficiency enhances Ca2+ influx and TMEM16F-mediated scrambling of PS on the plasma membrane
In platelets, PS exposure during coagulation or clearance is controlled by intracellular Ca2+ via Ca2+-activated scramblase and ion channels.1 As expected, A23187, a well-known Ca2+ ionophore, strongly induced PS exposure on the membranes of WT mouse platelets (Figure 4A). We examined the Ca2+ influx in platelets using the Fluo-4 AM calcium indicator and flow cytometry. As expected, the intracellular Ca2+ levels in SMS1-KO mouse platelets were significantly higher than those in WT mouse platelets (P = .0021) (Figure 4F; supplemental Figure 6), suggesting that reduced membrane SM due to SMS1 deficiency enhances Ca2+ influx and Ca2+-mediated PS exposure, leading to elevated platelet clearance and thrombocytopenia.
To elucidate the molecular mechanisms by which low SM levels affect Ca2+-dependent PS scrambling, we conducted in vitro cell experiments. Platelets are too small to use for cell biological experiments and do not grow in culture, and bone marrow–extracted MKs are difficult to use because of their limited amounts and inability to be passaged. We therefore used immortalized mouse embryonic fibroblasts (tMEFs) as a substitute. These cells were established previously from SMS-KO mice and were used for our SMS and SM function investigations.16,23 To confirm the elevation of plasma membrane PS exposure in SMS1-deficient cells, membrane PS levels were detected in WT, SMS1-KO, and SMS2-KO tMEFs. PS externalization to the outer leaflet was relatively high in only the SMS1-KO tMEFs; WT and SMS2-KO tMEFs had low PS exposure levels (Figure 5A). Next, the intracellular Ca2+ concentration was measured by using Fura2-AM and Ca2+ imaging. As with the higher PS exposure levels, the Ca2+ influx was also enhanced in SMS1-KO tMEFs compared with WT and SMS2-KO tMEFs (Figure 5B). These results indicate that SMS1 deficiency generally enhances PS exposure and Ca2+ influx in multiple cell types.
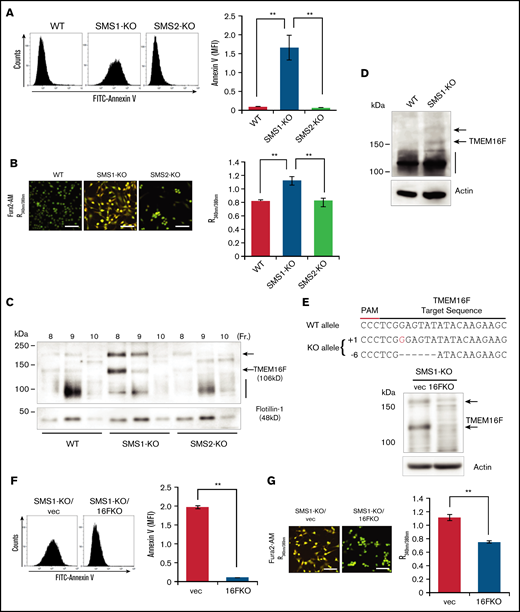
PS externalization and cytosolic Ca2+ level in tMEFs. (A) WT, SMS1-KO, and SMS2-KO tMEFs were stained with FITC–annexin V, and then analyzed with flow cytometry. The fluorescence of FITC–annexin V was quantified as mean fluorescent intensity (MFI). (B) Intracellular Ca2+ levels were measured with Fura2-AM and calcium imaging. The fluorescence was measured with Argus (aHamamtsu Photonics, Hamamatsu, Japan) and is presented as the 340 nm/380 nm fluorescence ratio (R340nm/380nm). Scale bars, 50 µm. (C) The DRM fraction was extracted with lysis buffer containing 1% Brij 58 (Sigma-Aldrich, St. Louis, MO) and OptiPrep (Axis-Shield Alere Technologies, Oslo, Norway) discontinuous gradients (5% and 30%). Fractions were collected from the top to the bottom of the gradient, and the DRM fractions (fractions 8-10) were analyzed by western blot analysis with antibodies against TMEM16F and the lipid raft marker flotillin-1. (D) Western blotting of TMEM16F protein in WT and SMS1-KO platelets was performed. (E) TMEM16F-KO tMEFs were established from SMS1-KO tMEFs by using CRISPER/Cas9 plasmids constructed based on lentiCRISPR-V2. The sequence of TMEM16F is shown at the top with the target sequence and the protospacer adjacent motif (PAM) sequence. Insertions are shown in red, and deletions are shown as black dashes. The change in length caused by each indel mutation is listed to the left of each sequence (+, insertion; −, deletion). TMEM16F protein in the SMS1-KO/TMEM16F-KO (1KO/16FKO) and SMS1-KO/vector (1KO/vec) tMEFs was detected by western blot analysis. Arrows indicate TMEM16F protein. (F) Cell surface PS was stained with FITC–annexin V and then analyzed with flow cytometry. The fluorescence was quantified as MFI. (G) Cytosolic Ca2+ levels were measured with Fura2-AM and are presented as the R340nm/380nm. Scale bars, 50 µm. Values show the mean ± standard deviation. **P < .005.
PS externalization and cytosolic Ca2+ level in tMEFs. (A) WT, SMS1-KO, and SMS2-KO tMEFs were stained with FITC–annexin V, and then analyzed with flow cytometry. The fluorescence of FITC–annexin V was quantified as mean fluorescent intensity (MFI). (B) Intracellular Ca2+ levels were measured with Fura2-AM and calcium imaging. The fluorescence was measured with Argus (aHamamtsu Photonics, Hamamatsu, Japan) and is presented as the 340 nm/380 nm fluorescence ratio (R340nm/380nm). Scale bars, 50 µm. (C) The DRM fraction was extracted with lysis buffer containing 1% Brij 58 (Sigma-Aldrich, St. Louis, MO) and OptiPrep (Axis-Shield Alere Technologies, Oslo, Norway) discontinuous gradients (5% and 30%). Fractions were collected from the top to the bottom of the gradient, and the DRM fractions (fractions 8-10) were analyzed by western blot analysis with antibodies against TMEM16F and the lipid raft marker flotillin-1. (D) Western blotting of TMEM16F protein in WT and SMS1-KO platelets was performed. (E) TMEM16F-KO tMEFs were established from SMS1-KO tMEFs by using CRISPER/Cas9 plasmids constructed based on lentiCRISPR-V2. The sequence of TMEM16F is shown at the top with the target sequence and the protospacer adjacent motif (PAM) sequence. Insertions are shown in red, and deletions are shown as black dashes. The change in length caused by each indel mutation is listed to the left of each sequence (+, insertion; −, deletion). TMEM16F protein in the SMS1-KO/TMEM16F-KO (1KO/16FKO) and SMS1-KO/vector (1KO/vec) tMEFs was detected by western blot analysis. Arrows indicate TMEM16F protein. (F) Cell surface PS was stained with FITC–annexin V and then analyzed with flow cytometry. The fluorescence was quantified as MFI. (G) Cytosolic Ca2+ levels were measured with Fura2-AM and are presented as the R340nm/380nm. Scale bars, 50 µm. Values show the mean ± standard deviation. **P < .005.
Because the binding of annexin V to PS is Ca2+-dependent, it is difficult to confirm whether the enhancement of PS exposure in SMS1-KO platelets is Ca2+-dependent via detection with annexin V. Therefore, we also used another PS-binding probe, lactadherin, to detect PS externalization in WT and SMS1-KO platelets.42 Confirming the findings shown in Figures 4A and 5A, PS externalization detected by fluorescein isothiocyanate (FITC)-conjugated lactadherin was increased in both platelets and tMEFs from SMS1-KO mice compared with those from WT mice (supplemental Figure 7). In addition, Ca2+ chelation by ethylene glycol tetraacetic acid suppressed this increased PS exposure in SMS1-KO mouse platelets and tMEFs, suggesting that the PS externalization promoted in SMS1-KO mice is Ca2+ dependent.
Previously, Gulshan et al17 showed that SM depletion inhibits PS flipping from outer leaflet to inner leaflet, leading to elevated PS exposure on the cell surface. We therefore examined flippase activity in tMEFs using nitrobenzoxadiazole (NBD)-labeled PS. However, there was no difference in the flipping activity level of NBD-labeled PS between WT and SMS1-KO tMEFs (supplemental Figure 8). In contrast, the uptake of NBD-labeled phosphatidylethanolamine was enhanced in SMS1-KO tMEFs compared with WT tMEFs. These results suggest that the increased PS exposure seen in SMS1-deficient mice is not due to an increased intracellular uptake of PS by flippase.
To investigate the role of SMS1-generated membrane SM on PS exposure and Ca2+ influx, we examined the changes in cell surface SM induced by bacterial sphingomyelinase (BSM) or exogenous C6-SM supplementation (supplemental Figure 9A,D).23 In WT tMEFs, BSM treatment decreased cell surface SM (supplemental Figure 9A). Along with the SM reduction, both PS exposure and Ca2+ influx were elevated in BSM-treated WT tMEFs (supplemental Figure 9B,C). Inversely, exogenous treatment of SMS1-KO tMEFs with C6-SM suppressed cell surface PS exposure and increased cell surface SM (supplemental Figure 9D,E). In addition, Ca2+ influx was also diminished in C6-SM–treated SMS1-KO tMEFs (supplemental Figure 9F). Similar results were observed in platelets; BSM treatment of WT mice cells increased the cell surface PS exposure (supplemental Figure 9G), whereas C6-SM supplementation reduced the PS externalization of SMS1-KO cells (supplemental Figure 9H). These data suggest that SMS1-generated cell surface SM is related to PS exposure and Ca2+ influx.
TMEM16F is the major Ca2+-dependent scramblase and ion channel that regulates PS exposure in platelets.9 However, decreased TMEM16F messenger RNA expression was not observed in either SMS1-KO platelets or tMEFs (supplemental Figure 10). Our previous studies showed that SMS1-generated SM is localized mainly in DRMs and regulates biological signaling, such as cell proliferation or cell migration, by affecting the responsiveness of receptors to their ligands.18 However, the effect of an SM-rich DRM on TMEM16F localization and activity is unknown. To investigate this, we determined the TMEM16F membrane composition in the DRM fractions of tMEFs. In WT and SMS2-KO tMEFs, TMEM16F was detected in the DRM fractions as an ∼106 kD protein (calculated molecular weight) (Figure 5C).9 In contrast, the TMEM16F in the DRM fractions of SMS1-KO tMEFs appeared larger (120 kD and 200 kD), possibly owing to glycosylation or aggregation.9 Concordantly, larger TMEM16F was observed in some SMS1-KO platelets (Figure 5D).
To examine the importance of the different TMEM16F size on PS exposure and Ca2+ influx, we performed a gene deletion of TMEM16F in SMS1-KO tMEFs by using the CRISPR/Cas9 system,43 establishing TMEM16F-KO/SMS1-KO (1KO/16FKO) tMEFs, which have a one-base increase in 1 allele and a six-base deletion in another allele of TMEM16F (Figure 5E). Depleted TMEM16F protein levels were observed in 1KO/16FKO tMEFs compared with control SMS1-KO tMEFs transduced with an empty vector (1KO/vec). The enhanced plasma membrane PS exposure observed in SMS1-KO tMEFs was not found in TMEM16F-depleted cells (Figure 5F). In addition, the elevated Ca2+ influx observed in other SMS1-KO cells was also suppressed in 1KO/16FKO tMEFs compared with 1KO/vec tMEFs (Figure 5G).
In addition to being regulated by TMEM16F, Ca2+ influx is affected by other mechanisms, such as T-type calcium channels and store-operated Ca2+ entry (SOCE).44 The T-type channels play a key role in remodeling Ca2+ homeostasis, and SOCE is a ubiquitous Ca2+ influx pathway that acts in response to Ca2+ store depletion of the endoplasmic reticulum. Neither the T-type calcium channel inhibitor pimozide nor the SOCE blocker YM58483 had any effect on the level of Ca2+ influx in SMS1-KO tMEFs (supplemental Figure 11). These results suggest that the increased cellular Ca2+ and PS externalization induced by SMS1 deficiency is caused by an acceleration of the Ca2+-dependent TMEM16F functions on DRMs.
Discussion
This study was initiated based on the present findings of decreased platelets and bleeding dysfunction in SMS1-KO mice but not SMS2-KO mice. In addition to reduced platelet numbers, we found the following pathological aspects of thrombocytopenia in SMS1-KO mice: (1) increased circulating reticulated platelets; (2) ectopic thrombopoiesis; and (3) splenomegaly. Further investigations revealed a significant PS exposure and subsequent clearance of platelets by phagocytes in the spleen, which was caused by the significant reduction of SM on the plasma membranes of platelets and MKs from SMS1-KO mice. SM-depleted membranes in SMS1-KO platelets/MKs induced an accelerated Ca2+ influx, causing PS externalization via a scramblase. Furthermore, SMS1 deficiency in tMEFs also enhanced PS exposure and Ca2+ influx and altered the behavior of the Ca2+-dependent scramblase TMEM16F in the DRM fraction. Interestingly, SMS1-KO tMEFs with a TMEM16F knockdown lacked not only the enhanced PS externalization but also the Ca2+ influx, suggesting that SMS1-generated SM-rich DRMs are a potential regulator of PS externalization, exerting their effects via two TMEM16F functions, as a phospholipid scramblase and a Ca2+-influx channel. We therefore concluded that SMS1-KO mice exhibit thrombocytopenia due to an enhanced platelet clearance through the activation of TMEM16F-mediated PS exposure on SM-depleted DRMs.
Homeostatic SM is synthesized by SMS1 and SMS2 in the Golgi apparatus and is delivered to organelle membranes (including the plasma membrane, mitochondrial membranes, and endosomal membranes), where it forms SM-rich DRMs, locations for responses to extracellular stimuli that house signaling receptors and membrane proteins and regulates diverse cellular functions such as proliferation, migration, and inflammation.18 However, the differences in how SMS1 and SMS2 affect SM-rich DRM formation are largely unclear. Our previous work using tMEFs revealed that both SMS1 and SMS2 contribute to the formation of SM-rich DRMs, which suppress cell migration via excessive CXCL12 responses.16 Similarly, serum-induced proliferation through an SM-rich membrane was also suppressed by knockdowns of either SMS1 or SMS2 in HeLa cells.45 In macrophages isolated from mice, SMS2 deficiency reduced membrane SM levels on DRMs and suppressed the inflammatory responses of tumor necrosis factor-α via preventing tumor necrosis factor receptor 1 recruitment to DRMs.46 In addition, the inflammatory response normally induced by lipopolysaccharide was suppressed in SMS1-KO macrophages.20 Notably, supplementation of membrane SM by SMS1 but not SMS2 was necessary for WR19L murine lymphoblast proliferation and survival through the recycling of transferrin/transferrin receptor complex.47 Moreover, T- and B-cell activations through their receptors were prevented by SM depletion of DRMs in SMS1-KO mice, resulting in SM-deficient amelioration of hepatitis or systemic lupus erythematosus, but not in SMS2-KO mice.19,48 Furthermore, JEV can achieve infection by attaching to target cell membranes through SMS1-generated SM.23 Consequently, SMS1-KO mice are protected from JEV infection and JEV-mediated encephalitis. Conversely, our recent report showed that SMS2, but not SMS1, contributes to the attachment and subsequent infiltration of lymphoma cells to mouse livers by supporting the expression of cell adhesion molecules such as ICAM-1 through membrane SM homeostasis.24 Therefore, the contributions of SM generation via SMS1 and SMS2 to various cell functions seem to depend on the cell/tissue and stimulation type. Here, SMS1, but not SMS2, was clearly shown to play a role in the homeostasis of PS distribution in the plasma membrane of platelets. Our collaboration with Kasahara et al25 revealed that SM levels on platelet DRMs decreased more in SMS1-KO mice than in SMS2-KO mice and that these levels are important for coagulation system function.
The mechanism by which the homeostasis of PS distribution in plasma membrane is maintained remains unknown. Many studies have found that alterations of membrane SM change the distributions of other lipids, including cholesterol and diacylglycerol. Nagao et al49 reported that depleting membrane SM boosted the ABCA1-mediated cholesterol efflux in Chinese hamster ovary cells and that supplementation with exogenous SM attenuated this phenomenon. Moreover, a regulatory effect of SM on the transbilayer movement of diacylglycerol in the plasma membrane was reported in Madin-Darby canine kidney cells as well as in a model membrane.50 Gulshan et al17 reported that, in addition to affecting cholesterol and diacylglycerol, SM increases the rate of spontaneous PS flipping, whereas the pharmacologic depletion of SM impairs PS inward transduction, leading to a subsequent PS increase on the outer leaflet. Our study similarly shows that SM depletion led to a significant appearance of PS on the plasma membrane outer leaflet in SMS1-KO platelets/MKs. In other blood cells, including lymphocytes and erythrocytes, lower levels of membranous SM were also confirmed in SMS1-KO mice compared with WT mice. However, because the PS elevations on the outer leaflet in these mice were clear but slight, corresponding erythrocytopenia and lymphocytopenia were not found in SMS1-KO mice (supplemental Figure 3). Therefore, SM generated by SMS1 is a potential regulator of PS content on the plasma membrane. Notably, we found that SMS1 deficiency had no effect on PS flipping from outer leaflet to inner leaflet (supplemental Figure 8), whereas PE flipping was enhanced in SMS1-deficient cells compared with WT cells; the implications of these findings are unknown, and further investigation is required.
We next explored additional mechanisms by which SM depletion induces PS exposure on the plasma membrane surface in SMS1-KO mice. The appearance of PS on the plasma membrane outer-leaflet is a well-known apoptosis sign for phagocytosis, acting as an “eat-me” signal in apoptotic cells.2 Similarly, platelets also undergo apoptosis after PS externalization and are cleared via phagocytosis in the spleen.4 Inhibition or deficiency of the antiapoptotic factor Bcl-xL, which blocks activation of the apoptotic factors Bak or Bax, enhances PS exposure and platelet clearance, leading to splenomegaly and thrombocytopenia.5,6 We found here that splenectomy of SMS1-KO mice recovered their decreased platelet numbers. In addition, the life span of SMS1-KO mouse platelets (t1/2 = 23 hours) was much shorter than that of WT mouse platelets (t1/2 = 62 hours; previous studies have reported similar WT mouse platelet half-lives [66 hours]).31,32 It is therefore suggested that platelet clearance is enhanced in SMS1-KO mice. Recently, it was reported that anti–glycoprotein Ib-IX antibody induces an enhancement of PS externalization through Akt activation in mouse platelets.39 The possible involvement of TMEM16F in anti-glycoprotein antibody-induced thrombocytopenia was also shown, although Nagata et al found that the coagulation impairment caused by TMEM16F deficiency acted through inhibiting PS externalization. Until now, there have been no reports showing a relationship between SMS1-dependent SM and TMEM16F-regulated PS externalization and Ca2+ influx.
Some PS externalization mechanisms, such as acting through the scramblases in platelet clearance, have been reported. Ca2+-dependent TMEM16F was shown to be the scramblase responsible for Scott syndrome, a bleeding disorder caused by dysfunctional PS externalization in platelets.9–12 Another PS membrane transporter is apoptosis-associated Xk-related protein 8 (Xkr8), which is a ten-transmembrane domain scramblase.51–53 In apoptosis induction, such as that following Bak or Bax activation, PS externalization on the plasma membrane is caused by Xkr8 scramblase.51 Similarly, in apoptotic platelets, TMEM16F-independent, Xkr8-mediated PS exposure was implicated in platelet activation and coagulation.52,53 Here, for the first time, we showed that Ca2+ influx and PS externalization, both of which are functions of TMEM16F, were enhanced in SMS1-deficient platelets and tMEFs. However, other apoptotic aspects, such as caspase activation and DNA fragmentation, were not observed in SMS1-KO cells (data not shown). In addition, SMS1-KO cells, such as tMEFs or WR19L cells, showed normal proliferation under experimental conditions.16,47 Xkr8 expression levels were significantly lower than TMEM16F expression levels in both tMEFs and platelets (supplemental Figure 10). We therefore focused on the roles of TMEM16F in Ca2+ influx and PS externalization and found that knocking out TMEM16F using the CRISPR/Cas9 system clearly suppressed both functions of TMEM16F, as an ion channel and a scramblase, in SMS1-KO tMEFs. Furthermore, the involvement of other Ca2+ influx mechanisms, such as T-type Ca2+ channel or SOCE, was not found by our experiments using specific inhibitors. Presently, because a relationship between Xkr8 and elevated PS externalization in SMS1-KO platelets cannot be completely ruled out, further investigation is needed.
The majority of adult ITP patients are treated with various therapies, such as the inhibition of autoantibody production or modulation of T-cell activity by steroids, reduction of autoantibody by immunoglobulins, reduction of platelet destruction by splenectomy, and the stimulation of platelet production by erythropoietin-like reagents. Steroid therapy is effective for two-thirds of ITP patients, but the other patients cannot be recovered from thrombocytopenia and progress to a refractory state. Therefore, some of them may have causes of thrombocytopenia other than immune dysfunction. Here, we report thrombocytopenia in SMS1-KO mice and show the involvement of sphingolipid SM regulated by SMS1 in the clearance of platelets, acting via the enhanced distribution of PS in plasma membrane outer leaflets through TMEM16F. In fact, PS externalization and Ca2+ influx in tMEFs were modified by the addition of BSM and C6-SM, inversely (supplemental Figure 9). Therefore, we strongly suggest that the regulation of SM production by SMS1 is a possible, novel therapeutic target for ITP, different from ongoing immune modulation.
Acknowledgments
The authors are grateful to Akira Hayashi, Shinji Aoki, and Hideo Ogiso (Kanazawa Medical University) for their excellent technical assistance. They also appreciate the kind gifts of Venus-lysenin from Toshihide Kobayshi (RIKEN) and of anti-TMEM16F polyclonal antibody from Shigekazu Nagata (Osaka University). The authors thank Edanz (https://jp.edanz.com/ac) for editing a draft of the manuscript.
This work was supported by grants from the JSPS KAKENHI (20K17666, Y.F.; 21K06827, M.T.; 18H02697, T.O.); the Takeda Science Foundation (2012); the Strategic Research Foundation Grant-aided Project for Private Universities, from MEXT (No. S1201004; H2012-16); and Kanazawa Medical University (H2012-15), 2012 to 2016.
Authorship
Contribution: S.N., Y.U., C.H., K.W., H.T, and T.K. performed research; Y.F. performed research, analyzed data, and wrote the manuscript; M.T. designed the study, performed research, analyzed data, supervised the study, and wrote the manuscript; and T.O. designed the study, supervised the study, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Toshiro Okazaki, Research Institute for Bioresources and Biotechnology, Ishikawa Prefectural University, 1-308 Suematsu, Nonoichi-shi, Ishikawa, 921-8836, Japan; e-mail: toshiroo@mbox.kyoto-inet.or.jp.

