

Key Points
CBFB-MYH11 transcripts and KIT mutations predict relapse in AML.
High-risk CBFB-MYH11 transcripts are associated with distinct transcriptional landscapes and upregulation of early hematopoiesis genes.
Introduction
Although the impacts of CBFB-MYH11 transcript variants on acute myeloid leukemia (AML) survival outcomes have been reported in adult patients with AML,1 this finding has not yet been tested in cohorts that include children and adolescents. Furthermore, the underlying reasons for these outcome differences have not been well studied. Therefore, we harnessed transcriptome sequencing performed on diagnostic AML samples of 1776 pediatric patients enrolled on front-line Children’s Oncology Group (COG) clinical trials. This comprehensive dataset includes 186 patients with CBFB-MYH11 fusions (supplemental Table 1; Figure 1A). Our analysis of these transcriptomes demonstrates that fusion transcript subtype predicts relapse in pediatric CBFB-MYH11 AML and deepens our understanding of how CBFB-MYH11 fusion transcripts may impact underlying leukemia biology and outcomes.
Methods
Patient samples and RNA-sequencing
AML samples were collected with informed consent from patients (0-28 years of age) enrolled on COG trials CCG-2961 (#NCT00002798),2 AAML03P1 (#NCT00070174),3 AAML0531 (#NCT00372593),4 and AAML1031 (#NCT01371981).5 Institutional review board approval for each protocol was obtained at each participating institution and submitted to the Cancer Trials Support Unit (CTSU) regulatory office. Each protocol was conducted in accordance with the Declaration of Helsinki. Total RNA derived from peripheral blood or bone marrow diagnostic specimens was purified using AllPrep DNA/RNA/miRNA Universal Kits. Purified RNA samples were then prepared for either strand-specific polyadenylated enriched messenger RNA libraries or strand-specific ribosome RNA-depleted libraries by the British Columbia Genome Sciences Center. Paired-end sequencing was performed on Illumina HiSeq 2000/2500 platforms, and sequence reads were aligned to the GRCh37 reference genome using BWA (v0.5.7).6 Reads were discarded based on mapping quality or if they failed the Illumina chastity filter, and duplicate reads were marked using Picard (v1.11). Gene level coverage analysis was performed using the British Columbia Genome Sciences Center pipeline v1.1 with Ensembl v69 annotations and was normalized based on RPKM or TPM.
Fusion calling
Fusion calls were made using CICERO, STAR-Fusion, and Trans-ABySS as previously described.7–9 In order to maximize detection sensitivity, CBFB-MYH11 transcript breakpoints called by one or more fusion callers were accepted after manual examination using Bambino.10 Overlapping calls between CICERO, STAR-Fusion, and Trans-ABySS for CBFB-MYH11 fusion transcripts were concordant at the level of exon transcript calling.
Variant calling
Variant calling was performed based on RNA sequencing and targeted DNA sequencing studies. RNA-seq libraries were constructed and sequenced as described above. All computational analyses were performed on a dedicated compute and storage infrastructure designed and implemented at St. Jude as previously described.11 Briefly, RNA reads were mapped using the StrongARM pipeline.12 Paired-end reads were aligned to 5 reference human genome databases (including GRCh37) using BWA,6 and the final BAM file was constructed by selecting the best of the 5 alignments. SNVs and Indels were called using Bambino10 and RNAIndel,13 followed by validation and pathogenicity classification as previously described.14,15
Statistical analysis
Event-free survival (EFS) and overall survival (OS) analysis was performed using Kaplan-Meier estimates. EFS was defined as the time from enrollment to first event (relapse, induction failure, or death) or last follow-up. OS was defined as the time from study enrollment to death or last follow-up. Relapse risk (RR) was defined as the time since end of Induction I to relapse or last follow-up. Cox proportional hazard regression models were employed to estimate hazard ratios for univariable and multivariable analyses of EFS, OS, and RR. Differences in EFS, OS, and RR between groups were determined using log-rank testing. The χ2 test was used to test the significance of observed differences in proportions, and Fisher’s exact test was used when data were sparse. Differences in medians were compared by the Mann-Whitney U test. A P value <.05 was considered statistically significant.
Gene expression analysis
Uniform manifold approximation and projection (UMAP) was performed using the uwot (v0.1.10) R package. Gene set enrichment analysis (GSEA) was performed using command line tools.16 In order to ensure a more uniform RNA-seq cohort, only ribosomal RNA-depleted sequenced libraries were included in the gene expression analysis.
Results and discussion
CBFB-MYH11 fusions in the 186 pediatric AML cases analyzed are predominantly composed of transcribed products from the fusion of exon 5 of CBFB with exon 33 of MYH11 (Figure 1A), hereafter referred to as E5-E33 CBFB-MYH11. To assess the prognostic significance of the E5-E33 transcript, we evaluated the probability of EFS, OS, and RR in our cohort stratified based on fusion transcript location (E5-E33 vs all others) (Figure 1B). Patients with E5-E33 had an EFS of 52% ± 8.1% (n = 156) at 5 years from diagnosis compared with an EFS of 80% ± 14.7% (n = 30) for those with other fusion transcripts (P value = .01).
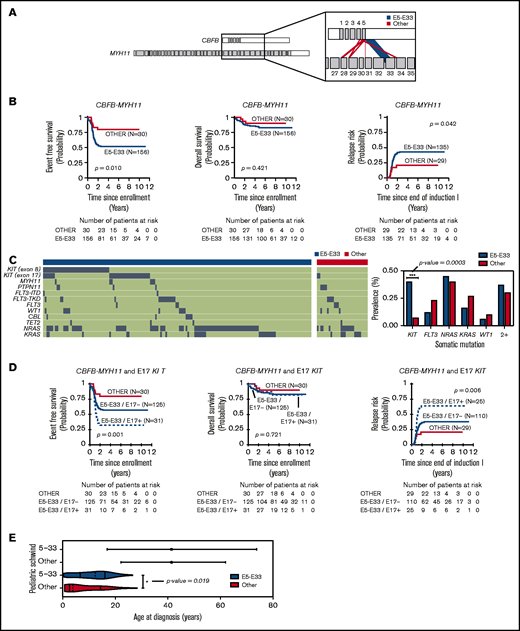
CBFB-MYH11 fusion transcript subtype predicts relapse in AML. (A) CBFB-MYH11 fusion transcripts within our cohort with line weights that correspond to the associated prevalence. (B) Kaplan-Meier estimates for the probability of EFS, OS, and RR in patients with CBFB-MYH11 AML stratified based on fusion transcript (E5-E33 vs all others). (C) Oncoplot with associated somatic driver mutations based on fusion transcript status. KIT mutations are more prevalent in E5-E33 CBFB-MYH11 AMLs compared with others (Fisher’s exact test, P value = .0003). (D) Kaplan-Meier estimates for the probability of EFS, OS, and RR in patients with CBFB-MYH11 AML stratified based on fusion transcript and E17 KIT mutation status. (E) Age distribution comparing Schwind et al (adults) and patients from our cohort (pediatrics).
CBFB-MYH11 fusion transcript subtype predicts relapse in AML. (A) CBFB-MYH11 fusion transcripts within our cohort with line weights that correspond to the associated prevalence. (B) Kaplan-Meier estimates for the probability of EFS, OS, and RR in patients with CBFB-MYH11 AML stratified based on fusion transcript (E5-E33 vs all others). (C) Oncoplot with associated somatic driver mutations based on fusion transcript status. KIT mutations are more prevalent in E5-E33 CBFB-MYH11 AMLs compared with others (Fisher’s exact test, P value = .0003). (D) Kaplan-Meier estimates for the probability of EFS, OS, and RR in patients with CBFB-MYH11 AML stratified based on fusion transcript and E17 KIT mutation status. (E) Age distribution comparing Schwind et al (adults) and patients from our cohort (pediatrics).
We next harnessed variant calls on the same cohort of patients (n = 186) to elucidate the mutational landscape of CBFB-MYH11 AMLs. KIT mutations were significantly enriched in E5-E33 CBFB-MYH11 AMLs with a prevalence of 42% in the E5-E33 cohort vs 7% in the Other cohort (P value = .0003; Figure 1C; supplemental Table 1). In contrast, the prevalence of FLT3, NRAS, KRAS, and WT1 mutations was not significantly different between the 2 groups (Figure 1C). We have previously demonstrated that exon 17 (E17) KIT mutations are associated with adverse outcomes in patients with core-binding factor AML.1,17 Therefore, we assessed for their impact on survival within our E5-E33 cohort and found that they were strongly associated with a significantly worse EFS of 32% ± 16.8% for E17 KIT+ at 5 years from diagnosis compared with an EFS of 56% ± 10.5% for E17 KIT− patients (P value = .042) (Figure 1D) and was notably independent of E17 KIT variant allele frequency (supplemental Figure 1). Conversely, exon 8 (E8) KIT mutations were associated with no difference in survival (supplemental Figure 2).
The prognostic impact of E5-E33 transcripts on EFS was maintained in multivariable survival analysis that included age, white blood cell count, and E17 KIT mutations (supplemental Tables 2 and 3) and in survival analysis that excluded KIT mutant AMLs (supplemental Figure 3). Furthermore, the inferior outcomes associated with E5-E33 CBFB-MYH11 AMLs that are significantly worse in the presence of an additional E17 KIT mutation are particularly relevant because they address a gap in knowledge based on previously studied demographic age groups (Figure 1E) and further support the hypothesis that CBFB-MYH11 transcript subtypes impact leukemia biology and/or chemoresistance.
Interestingly, and in contrast to Schwind et al,1 despite adverse EFS in E5-E33 patients, OS did not differ based on fusion transcript (Figure 1B,D) suggesting that relapses in children, adolescents, and young adults with E5-E33 CBFB-MYH11 AMLs ± E17 KIT mutations are highly salvageable. Because hematopoietic stem cell transplantation (HSCT) in first complete remission is deferred to after an initial relapse in favorable risk pediatric patients (with only 1 of 186 patients within our cohort undergoing HSCT in first complete remission), we hypothesize that leukemia cells with CBFB-MYH11 fusion oncogene are highly susceptible to the allogeneic effect of the HSCT.
Next, we performed unbiased GSEA comparing E5-E33 CBFB-MYH11 AMLs with all others with alternative breakpoints across the Molecular Signature Database18,19 (specifically, the hallmark, curated, and oncogenic signature gene sets). We excluded KIT mutant AMLs because they have been shown to be associated with a distinct gene expression profile.20 Intriguingly, 3 previously published gene sets that established and defined CBFB-MYH11 AML transcriptional signatures are significantly enriched in E5-E33 CBFB-MYH11 AMLs compared with non–E5-E33 CBFB-MYH11 AMLs (Figure 2A), suggesting that non–E5-E33 CBFB-MYH11 AMLs represent a distinct transcriptional subtype. Indeed, performing UMAP analysis revealed that E5-E33 CBFB-MYH11 AMLs occupy a distinct cluster from most other CBFB-MYH11 AMLs (P < .001), which are more heterogeneous in their transcriptional clustering (Figure 2B).
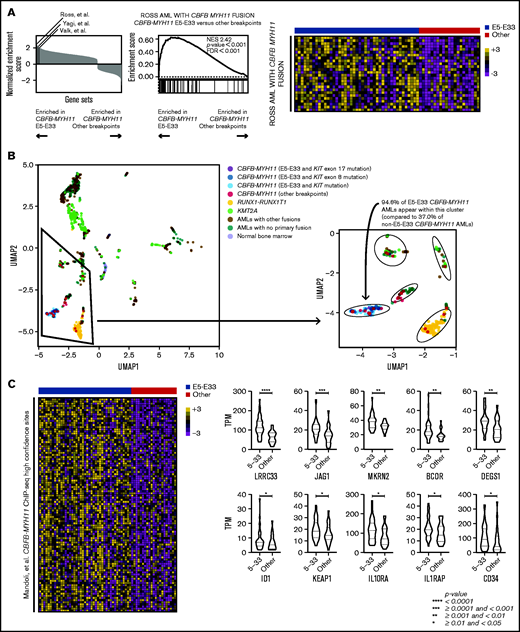
CBFB-MYH11 fusion transcript subtypes are associated with distinct transcriptional landscapes. (A) GSEA comparing E5-E33 CBFB-MYH11 AMLs with all others, using gene sets available through the Molecular Signature Database (hallmark, curated, and oncogenic signature gene sets). Three gene sets that define CBFB-MYH11 transcriptional signatures are significantly enriched in E5-E33 CBFB-MYH11 AMLs. Specifically, the Ross et al gene set is associated with a normalized enrichment score of 2.42 (P value <.0001) with the displayed heatmap of enriched genes. (B) UMAP analysis revealed that E5-E33 CBFB-MYH11 AMLs occupy a different cluster than most other CBFB-MYH11 AMLs. (C) GSEA comparing E5-E33 CBFB-MYH11 AMLs with all others and using a gene set derived by Mandoli et al based on CBFB-MYH11 chromatin immunoprecipitation sequencing (ChIP-seq) occupancy reveals significant enrichment (normalized enrichment score of 1.37, P value <.0001) in E5-E33 CBFB-MYH11 AMLs (100 most enriched genes shown). Violin plots of representative genes within the Mandoli et al gene set that are associated with hematopoietic stem cells or early hematopoiesis (Mann-Whitney U test).
CBFB-MYH11 fusion transcript subtypes are associated with distinct transcriptional landscapes. (A) GSEA comparing E5-E33 CBFB-MYH11 AMLs with all others, using gene sets available through the Molecular Signature Database (hallmark, curated, and oncogenic signature gene sets). Three gene sets that define CBFB-MYH11 transcriptional signatures are significantly enriched in E5-E33 CBFB-MYH11 AMLs. Specifically, the Ross et al gene set is associated with a normalized enrichment score of 2.42 (P value <.0001) with the displayed heatmap of enriched genes. (B) UMAP analysis revealed that E5-E33 CBFB-MYH11 AMLs occupy a different cluster than most other CBFB-MYH11 AMLs. (C) GSEA comparing E5-E33 CBFB-MYH11 AMLs with all others and using a gene set derived by Mandoli et al based on CBFB-MYH11 chromatin immunoprecipitation sequencing (ChIP-seq) occupancy reveals significant enrichment (normalized enrichment score of 1.37, P value <.0001) in E5-E33 CBFB-MYH11 AMLs (100 most enriched genes shown). Violin plots of representative genes within the Mandoli et al gene set that are associated with hematopoietic stem cells or early hematopoiesis (Mann-Whitney U test).
To uncover candidate mechanisms to explain the differences in survival outcomes and transcriptional landscapes between E5-E33 CBFB-MYH11 AMLs and others, we analyzed our transcriptome data in the context of established CBFB-MYH11 ChIP-seq experimental data.21 We found that E5-E33 CBFB-MYH11 AMLs were enriched in the gene set that encompasses all genes located near high-confidence CBFB-MYH11 ChIP-seq sites (Figure 2C), which included genes implicated in hematopoietic stem cell self-renewal, such as LRRC33,22 JAG1,23 MKRN2,24 BCOR,25 DEGS1,26 ID1,27 KEAP1,28 IL10RA,29 IL1RAP,30 and CD34.31–35 On the basis of their locations in close proximity with experimentally validated CBFB-MYH11 ChIP-seq sites and their differential expression based on CBFB-MYH11 transcript subtypes, these genes represent candidate transcriptional markers associated with inferior survival outcomes and present an opportunity for further functional validation and/or therapeutic targeting studies.
Acknowledgments
The authors thank the Library Construction, Biospecimen, Sequencing, and Bioinformatics teams at Canada’s Michael Smith Genome Sciences Centre for expert technical assistance.
This work was supported by grants from the Rally Foundation, Rally Foundation Career Development Award (B.J.H.), the St. Baldrick's Foundation, St. Baldrick's Scholar with generous support from RowOn 4 A Cure (B.J.H.), St. Baldrick's Consortium Grant (S.M.), Target Pediatric AML (S.M.), Leukemia and Lymphoma Society 6558-18 (S.M., E.A.K), National Institutes of Health, National Cancer Institute R01-CA114563-10 (S.M.), HHSN-261200800001E (S.M.), National Cancer Institute COG Chair U10-CA098543 (S.M.), Andrew McDonough B+ Foundation (S.M.), Frank A. Campini Foundation (B.J.H.), Hyundai Hope on Wheels (B.J.H. and S.M.), Fund for Innovation in Cancer Informatics (X.M.), National Cancer Institute NCTN Statistics & Data Center U10-CA180899 (S.M., T.A.A.), National Cancer Institute NCTN Operations Center Grant U10-CA180886 (S.M., E.A.K.), Project Stella (S.M.).
This work used the computational infrastructure of Fred Hutchinson Cancer Research Center (FHCRC) Scientific Computing funded by ORIP grant S10OD028685.
The results published here are in part based on data generated under the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project managed by the National Cancer Institute. Information about TARGET can be found at https://ocg.cancer.gov/.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This paper is a report from the Children’s Oncology Group.
Authorship
Contribution: B.J.H., X.M., and S.M. conceived and designed the research; J.L.S., Y.-C.W., A.R.L., R.E.R., R.G., and T.A.A. collected and assembled the data; B.J.H., J.L.S., Y.-C.W., K. Taghizadeh., A.R.L., R.E.R., Y.L., P.K., R.G., T.A.A., X.M., and S.M. analyzed and interpreted the data; all authors wrote the paper, had final approval of manuscript, and were accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Benjamin J. Huang, University of California San Francisco, 550 16th Street, Box 0434, San Francisco, CA 94158; e-mail: ben.huang@ucsf.edu.

