

Key Points
Typical features of GATA2 deficiency were associated with a de novo tandem duplication of GATA2 and increased expression of GATA2-AS1.
The duplication contained a maternally inherited deletion copy number polymorphism esv2725896/nsv513733.
Abstract
A 3-year-old girl of nonconsanguineous healthy parents presented with cervical and mediastinal lymphadenopathy due to Mycobacterium fortuitum infection. Routine blood analysis showed normal hemoglobin, neutrophils, and platelets but profound mononuclear cell deficiency (monocytes < 0.1 × 109/L; B cells 78/μL; NK cells 48/μL). A 548 902-bp region containing GATA2 was sequenced by targeted capture and deep sequencing. This revealed a de novo 187-kb duplication of the entire GATA2 locus, containing a maternally inherited copy number variation deletion of 25 kb (GRCh37: esv2725896 and nsv513733). Many GATA2-associated phenotypes have been attributed to amino acid substitution, frameshift/deletion, loss of intronic enhancer function, or aberrant splicing. Gene deletion has been described, but other structural variation has not been reported in the germline configuration. In this case, duplication of the GATA2 locus was paradoxically associated with skewed diminished expression of GATA2 messenger RNA and loss of GATA2 protein. Chimeric RNA fusion transcripts were not detected. A possible mechanism involves increased transcription of the anti-sense long noncoding RNA GATA2-AS1 (RP11-472.220), which was increased several fold. This case further highlights that evaluation of the allele count is essential in any case of suspected GATA2-related syndrome.
Introduction
GATA2 is expressed in early hematopoietic progenitors and is required for stem cell longevity and lympho-myeloid differentiation.1 Germline heterozygous mutation of GATA2 is associated with progressive loss of mononuclear cell production, immunodeficiency, myelodysplasia, familial acute myeloid leukemia, lymphedema, and deafness.2-5 Hundreds of cases associated with sequence variation in the GATA2 gene causing amino acid substitution, frameshift/deletion, loss of intronic enhancer function, or aberrant splicing have been described.6-10 Structural variation is less common, although several cases of germline gene deletion have been described,11 and displacement of the distal enhancer by inv(3)(q21;q26) or t(3;3)(q21;q26) is well documented in high-risk leukemia.12 Here, we describe a patient presenting with typical features of mycobacterial infection associated with severe monocytopenia, B-cell and NK cell lymphopenia, and myelodysplasia who had a de novo 187-kb duplication of the entire GATA2 locus associated with skewed diminished expression of GATA2, loss of GATA2 protein, and increased transcription of the anti-sense long noncoding RNA GATA2-AS1 (RP11-472.220).
Case description
A 3-year old girl of nonconsanguineous healthy parents presented with cervical and mediastinal lymphadenopathy due to Mycobacterium fortuitum infection (2010). Her parents reported that she had hearing difficulties. Routine blood analysis showed normal hemoglobin, neutrophils, and platelets but profound mononuclear cell deficiency (monocytes < 0.1 × 109/L; B cells 78/μL; and NK cells 48/μL). Immunoglobulins were normal (IgG, 11.3 g/dL; IgA, 0.95 g/dL, IgM, 1.72 g/dL), but mitotic responses of whole blood were diminished. She was treated with ciprofloxacin, amikacin, sulfamethoxazole, and linezolid but did not respond and required surgical intervention for airway compression due to mediastinal lymphadenopathy. Sensorineural deafness was documented after antibiotic therapy. She was subsequently treated with interferon-γ and her condition slowly improved. However, by the age of 8 years she developed a chronic respiratory illness with persistent cough, fatigue, nodular and ground glass infiltrates, and reduced diffusion capacity. Lung biopsy showed peribronchiolar infiltrates, but no evidence of infection or pulmonary alveolar proteinosis. Bone marrow examination revealed multi-lineage dysplasia with trisomy 8 in 5 of 22 metaphases. She underwent successful allogeneic bone marrow transplantation using a matched unrelated donor at age 9 years. Four years later she has normal cell counts and remains free of infections and respiratory compromise. Genetic screening was negative for mutations of IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, ISG15, and IRF8.13 No mutations of GATA2 were detected in coding regions or enhancers, but MLPA suggested the existence of a whole gene duplication. Fms-like tyrosine kinase-3 (Flt3) ligand was markedly elevated (7940 pg/mL).
Methods
Patient material was obtained with informed consent through the French GATA2-like Project and assigned to Newcastle Biobank by material transfer agreement (Newcastle and North Tyneside 1 Research Ethics Committee Reference 17/NE/0361). Whole-blood DNA of the patient, both parents, and 2 unrelated healthy controls was extracted using a QIAamp DNA Mini-Kit (QIAGEN). A 548 902-bp region containing GATA2, flanked by EEFSEC and RPN1, was obtained by custom capture with Nimblegen baits for deep sequencing on Illumina platforms (98% coverage ≥ 1000 fold) (Figure 1A). Sequence was aligned to GRCh37 using Burrows-Wheeler Aligner.14 Structural variation was analyzed with ONCOCNV v6.8,15 using the parents and healthy controls as baseline and by manual inspection of discordant read pairs and soft-clipped reads in Integrative Genomics Viewer.16 GATA2 protein was detected by western blot using rabbit polyclonal antibody (Abcam; ab153820). Allelic expression was assessed in cDNA amplified with CCCGGGACTGGTGCTCTTTC/GGCAGAAACCCTGTGGGTCC (rs1573858); CCCTCTAGAGCCCTGTAGTTCCT/TGAGCGGGGT TGGCATAGTAG (rs2335237). Stranded messenger RNA sequencing was performed with Illumina NextSeq to obtain 150-bp paired-end reads at 134 to 155 million reads per sample on libraries prepared with a SmartSeq Kit 2 (Takara) from RNA extracted with a QIAamp RNA Micro Kit (QIAGEN). Reads were aligned with STAR17 and examined for fusions with Arriba.18
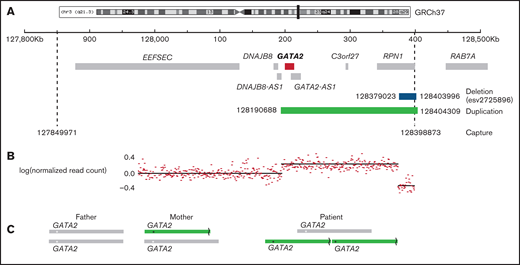
Region of chromosome 3 showing copy number variation in the patient. (A) The captured region (3:127849971-128398873) is indicated by dashed lines, the duplication detected (3:128190688-128404309) is shown in green, and the deletion (3:128379023-128303996) is shown in blue. (B) The normalized read count mapping to the captured region. (C) The predicted arrangement of alleles in the parents and patient. The asterisk (*) indicates the position of GATA2, and the broken line denotes the site of the deletion.
Region of chromosome 3 showing copy number variation in the patient. (A) The captured region (3:127849971-128398873) is indicated by dashed lines, the duplication detected (3:128190688-128404309) is shown in green, and the deletion (3:128379023-128303996) is shown in blue. (B) The normalized read count mapping to the captured region. (C) The predicted arrangement of alleles in the parents and patient. The asterisk (*) indicates the position of GATA2, and the broken line denotes the site of the deletion.
Results and discussion
The patient had a de novo tandem duplication of 187 kb spanning GATA2 and RPN1. The duplication, which has not been previously reported, contained a deletion of 25 kb 5' of RPN1. The deletion was inherited from the mother (Figure 1) and is a relatively common copy number variation reported in 4% of Europeans (GRCh37: esv2725896 and nsv513733).19 The prevalence of deletions in this region indicates that it is susceptible to structural variation. However, the deletion alone has no reported phenotype, even though it removes an alternative transcription start site for RPN1 at 128 400 kb, associated with CTCF and H3K27ac binding peaks. A second copy of GATA2 was joined to this region by the tandem duplication. It was not obvious why duplication of the GATA2 region should result in a classical GATA2-deficiency phenotype. GATA2 was not detected in patient peripheral blood mononuclear cells (PBMCs) or healthy control PBMCs by western blot. However, a consistent reduction was observed in the patient using primary fibroblast lines, in which GATA2 is easily detected (Figure 2A). Analysis of heterozygous single-nucleotide polymorphisms indicated skewed expression toward the maternal allele, relative to genomic sequence chromatograms, in PBMCs and fibroblast cDNA (Figure 2B), and overall expression was diminished in 3 of 4 amplicons tested (Figure 2C). Unexpectedly, the expression of GATA2-AS1 was increased in patient PBMCs and fibroblasts by quantitative PCR and RNA sequencing (Figure 2C-D). This long noncoding RNA, also known as RP11-472.22, is widely expressed and associated with MYC-induced loss of GATA2 in several cancers.20-22 The tandem duplication brings the second copy of GATA2 much closer to its distal enhancer, potentially activating transcription of the locus. It is not clear why GATA2-AS1 predominates and appears to inhibit transcription of all 3 copies of the GATA2 gene, including the paternal allele in trans (Figure 2E). The mechanism of skewed expression of the duplicated maternal allele is not clear, although suppression of a wild-type allele has also been reported with GATA2 T354M substitution,23 and monoallelic expression is observed in sporadic leukemia.24 High-depth RNA sequencing of PBMCs and fibroblasts did not reveal any novel fusion transcripts (eg, chimeric RPN1-GATA2; supplemental Table 1).
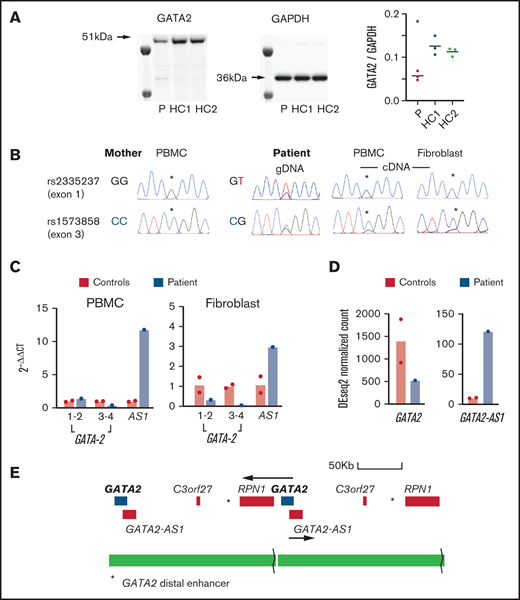
Expression of GATA2 and GATA2-AS. (A) Western blots of GATA2 and GAPDH in fibroblasts from the patient (P) and 2 healthy controls (HC1, HC2) showing ∼50% reduction in the ratio of GATA2/GAPDH. Samples were loaded in triplicate for quantification as shown. Three independent experiments were performed with similar results. *P < .05 vs control. (B) Allelic expression was investigated by preparing genomic DNA (gDNA) and complementary DNA (cDNA) from maternal PBMCs, patient PBMCs, and fibroblasts. Two single-nucleotide polymorphisms rs2335237 (exon 1) and rs1573858 (exon 3), heterozygous in the patient and homozygous in the mother, showed preferential expression of the duplicated maternal allele above the peak heights observed in the genomic DNA sequence chromatogram (asterisks). (C) Quantitative polymerase chain reaction analysis of GATA2 expression in PBMCs and fibroblasts comparing ΔΔCt (GATA2-GAPDH) in 2 controls and the patient using 3 primer pairs for GATA2 exons 1-2, exons 3-4, and the GATA2 anti-sense transcript (AS1). (D) GATA2 and GATA2-AS1 expression determined from RNA sequencing data in PBMCs from the patient and 2 controls. (E) Structure of the duplication showing the position of the GATA2 distal enhancer (asterisk) in relation to the second copy of GATA2 and the potential hybrid transcript of GATA2 and RPN1.
Expression of GATA2 and GATA2-AS. (A) Western blots of GATA2 and GAPDH in fibroblasts from the patient (P) and 2 healthy controls (HC1, HC2) showing ∼50% reduction in the ratio of GATA2/GAPDH. Samples were loaded in triplicate for quantification as shown. Three independent experiments were performed with similar results. *P < .05 vs control. (B) Allelic expression was investigated by preparing genomic DNA (gDNA) and complementary DNA (cDNA) from maternal PBMCs, patient PBMCs, and fibroblasts. Two single-nucleotide polymorphisms rs2335237 (exon 1) and rs1573858 (exon 3), heterozygous in the patient and homozygous in the mother, showed preferential expression of the duplicated maternal allele above the peak heights observed in the genomic DNA sequence chromatogram (asterisks). (C) Quantitative polymerase chain reaction analysis of GATA2 expression in PBMCs and fibroblasts comparing ΔΔCt (GATA2-GAPDH) in 2 controls and the patient using 3 primer pairs for GATA2 exons 1-2, exons 3-4, and the GATA2 anti-sense transcript (AS1). (D) GATA2 and GATA2-AS1 expression determined from RNA sequencing data in PBMCs from the patient and 2 controls. (E) Structure of the duplication showing the position of the GATA2 distal enhancer (asterisk) in relation to the second copy of GATA2 and the potential hybrid transcript of GATA2 and RPN1.
The physiological function of GATA2 is critically dependent upon its level of expression, and it is known that overexpression in the progenitor compartment can disrupt hematopoiesis.25 We did not have access to bone marrow progenitors to exclude this possibility, although the phenotypic similarity with known GATA2 deficiency suggests that loss of expression is the mechanism.
In summary, this case illustrates that structural variation and epigenetic mechanisms potentially involving GATA2-AS1 can lead to constitutive loss of GATA2 expression and a classical GATA2-deficiency phenotype. The clinical presentation of such a case remains a strong argument to avoid transplantation from a related donor, given the possibility of an inherited genetic defect. The results further emphasize that it is important to evaluate the number of GATA2 alleles in suspected cases of GATA2-related syndrome.
Acknowledgments
P.S. and M.C. received funding from UKRI (MRC) project grant MR/P002005/1. M.H. was funded by an EPSRC DTP studentship. D.R. received a Wellcome Trust Seed Award in Science (206103/Z/17/Z). The authors gratefully acknowledge the Association Laurette Fuguain and Bright Red for additional contributions to funding.
Authorship
Contribution: P.S., M.R., L.L., S.D., N.P., R.E.D., and B.N. performed research; M.H., A.R., and A.M. analyzed and interpreted data; J.B. designed research and collected data; V.B. designed research and interpreted data; E.D. and D.R. designed research and analyzed and interpreted data; M.P. designed research and collected, analyzed, and interpreted data; and M.C. designed research, analyzed and interpreted data, and wrote the manuscript
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew Collin, Newcastle University, Framington Pl, Newcastle upon Tyne NE2 4HH, United Kingdom; e-mail: matthew.collin@ncl.ac.uk.

