

Key Points
Skin-restricted PCFCL is genetically distinct from usual FL and cutaneous FL with concurrent or future systemic involvement.
We propose 3 criteria based on molecular features to predict cases of cutaneous FL with concurrent or future systemic involvement.

Abstract
Primary cutaneous follicle center lymphomas (PCFCLs) are indolent B-cell lymphomas that predominantly remain skin restricted and manageable with skin-directed therapy. Conversely, secondary cutaneous involvement by usual systemic follicular lymphoma (secondary cutaneous follicular lymphoma [SCFL]) has a worse prognosis and often necessitates systemic therapy. Unfortunately, no histopathologic or genetic features reliably differentiate PCFCL from SCFL at diagnosis. Imaging may miss low-burden internal disease in some cases of SCFLs, leading to misclassification as PCFCL. Whereas usual systemic FL is well characterized genetically, the genomic landscapes of PCFCL and SCFL are unknown. Herein, we analyzed clinicopathologic and immunophenotypic data from 30 cases of PCFCL and 10 of SCFL and performed whole-exome sequencing on 18 specimens of PCFCL and 6 of SCFL. During a median follow-up of 7 years, 26 (87%) of the PCFCLs remained skin restricted. In the remaining 4 cases, systemic disease developed within 3 years of diagnosis. Although the SCFLs universally expressed BCL2 and had BCL2 rearrangements, 73% of the PCFCLs lacked BCL2 expression, and only 8% of skin-restricted PCFCLs had BCL2 rearrangements. SCFLs showed low proliferation fractions, whereas 75% of PCFCLs had proliferation fractions >30%. Of the SCFLs, 67% had characteristic loss-of-function CREBBP or KMT2D mutations vs none in skin-restricted PCFCL. Both SCFL and skin-restricted PCFCL showed frequent TNFRSF14 loss-of-function mutations and copy number loss at chromosome 1p36. These data together establish PCFCL as a unique entity with biological features distinct from usual systemic FL and SCFL. We propose 3 criteria based on BCL2 rearrangement, chromatin-modifying gene mutations (CREBBP, KMT2D, EZH2, and EP300), and proliferation index to classify cutaneous FL specimens based on the likelihood of concurrent or future systemic spread.
Introduction
Primary cutaneous follicle center lymphoma (PCFCL) is the most common type of primary cutaneous B-cell lymphoma, representing ∼60% of cases. PCFCLs are characterized by cutaneous proliferation of clonal germinal center lymphocytes, “follicle center cells,” that most frequently present as plaques and nodules on the scalp, forehead, or trunk. By definition, they occur in the absence of nodal or other extracutaneous involvement at the time of diagnosis. Most cases remain localized to the skin and are remarkably indolent, with a 5-year disease-specific survival of 95%, despite a high recurrence rate in the skin.1,2 Historically, it has been challenging to distinguish these cases of PCFCL with indolent behavior from the more aggressive cutaneous lymphomas. In fact, PCFCLs with diffuse growth patterns were often classified as diffuse large B-cell lymphoma, resulting in overtreatment of PCFCL with multiagent chemotherapy, until the World Health Organization-European Organization for Research and Treatment of Cancer classification in 2005 recognized that PCFCL had a morphological spectrum that included follicular, follicular and diffuse pattern, and diffuse pattern. Clinically, it is critically important to distinguish PCFCL from cutaneous involvement by systemic follicular lymphomas (ie, secondary cutaneous follicular lymphomas [SCFLs]) which are largely incurable, relapse more frequently, generally have a worse prognosis, and are managed differently.1,3,4
Despite numerous studies, there are currently no reliable clinicopathologic or molecular criteria to distinguish PCFCL from SCFL. In contrast to SCFL, cases of PCFCL usually lack BCL2 expression, often have weak or negative CD10 expression, and may lack BCL2 gene rearrangements.5,6 Although these features have been helpful in distinguishing PCFCL from SCFL, none is specific, as up to 30% of cases of PCFCL have BCL2 gene rearrangements and/or express BCL2 or CD10.3,4,7-10 As a result, imaging is relied on to assess for the possibility of systemic involvement. Although imaging successfully identifies concurrent systemic involvement in many cases, its absolute sensitivity is not clear. Interestingly, ∼10% of PCFCLs later disseminate extracutaneously.11 It is currently unknown whether these cases are genetically more similar to skin-restricted PCFCL or to SCFL. The latter would suggest that these cases may actually represent SCFL in which systemic disease is present but below the level of detection at initial diagnosis and is thus misdiagnosed as PCFCL. To date, there are no available histopathologic or genetic features to reliably identify cutaneous FL without concurrent systemic involvement that later disseminates beyond the skin.
The genetic landscape of usual systemic FL is well-characterized,12-24 but the genetic landscape of PCFCLs remains unclear. Emerging data suggest that other FL subtypes have distinct genetics. For example, pediatric-type follicular lymphoma (PTFL), a rare variant of localized nodal FL that occurs primarily in younger patients, is characterized by TNFRSF14, IRF8, and activated MAPK pathway mutations.1-3,25,26
Herein, we describe the genomic landscapes of PCFCLs and SCFLs and define the genetic similarities and differences between cutaneous FLs and other FL subtypes. Based on these molecular differences, we propose 3 criteria to determine whether a cutaneous follicular lymphoma is likely to spread systemically.
Materials and methods
Sample preparation and sequencing analysis
All studies were approved by the Institutional Review Boards of Northwestern University and Massachusetts General Hospital. Deidentified formalin-fixed, paraffin-embedded (FFPE) archival specimens from 40 cutaneous FLs were collected from the Medical University of Graz (n = 8), Northwestern University (NU; n = 2), Massachusetts General Hospital (MGH; n = 25), and Brigham and Women’s Hospital (BWH; n = 5) and were reviewed by expert dermatopathologists (J.G. and L.C.) and/or a hematopathologist (A.L.). Cases of PCFCL and SCFL were categorized based on clinical history. PCFCL is first diagnosed in the skin and has no systemic involvement at the time of diagnosis, and SCFL has either concurrent systemic or preceding systemic disease. Genomic DNA was prepared, sequenced, and analyzed as previously described.27 Median coverage depth was >150 independent reads per targeted base. Putative driver genes were identified as has been described.27
Copy number aberrations
Genomic DNA was processed for molecular inversion probe array analysis with the OncoScan FFPE Assay kit (Thermo Fisher, Santa Clara, CA), as described.28 Data analysis was performed with Chromosome Analysis software (ChAS), version 3.2 (Thermo Fisher), and Nexus Express Software for OncoScan, version 3.1 (BioDiscovery, Hawthorne, CA), with reference to assembly GRCh37/hg19, by a published method.29 All cases were processed with the TuScan segmentation algorithm (ChAS; Thermo Fisher), except for case PS02, which was recentered and processed by using SNP-FASST2 (OncoScan; Nexus Express). Recurrent genomic alterations were calculated with the aggregate analysis in Nexus Express.
Whole-exome sequencing
Genomic DNA from 18 cases of PCFCL and 6 of SCFL was prepared, sequenced, and analyzed as previously described.26,27,30,31 Whole-exome capture was performed with the Agilent SureSelect Human All Exon v5 (Agilent Technologies) and xGen Exome Research Panel (Integrated DNA Technologies) bait sets. In summary, genomic DNA was sheared, end repaired, ligated with barcoded Illumina sequencing adapters, amplified, size selected, and captured with the bait sets. The resulting Illumina exome-sequencing libraries were then quantified by quantitative polymerase chain reaction, pooled, and sequenced with 76-bp paired-end reads, using Illumina HiSeq 2000, 3000, or 4000 sequencers. Coverage depth was >150 independent reads per targeted base. Sequences were aligned to the hg19/GRCh37 build of the reference human genome sequence and annotated with ELAND (Illumina) or Oncotator (Broad Institute).
Identification of somatic variants and putative driver genes
Significantly mutated genes were identified with our multitiered pipeline, which identifies cancer-promoting mutations that occur more often than expected by chance.27,30 In addition, we examined our data set for damaging mutations (for putative tumor suppressors) and for recurrent amino acid alterations (for putative oncogenes). We first examined our PCFCL and SCFL cohorts, a cohort of published B-cell lymphomas, and the Catalogue of Somatic Mutations in Cancer (COSMIC).32 We looked for mutations in our PCFCL and SCFL data sets that are also found in COSMIC hotspots (>20 mutations within 5 amino acids of the designated mutation position), recurrent mutations in published B-cell lymphomas, and damaging mutations in canonical tumor suppressors.
Comparison of PCFCL and SCFL with other FL subtypes
We compared the frequency of mutations in genes most commonly mutated in defined types of FL (usual systemic FL, diffuse FL, and PTFL) with that of the same genes in PCFCL or SCFL. We normalized these values to a 0 to 1 similarity index scale, where 1 was exact correlation and 0 was no correlation.
Immunohistochemical staining and fluorescence in situ hybridization
Immunohistochemical (IHC) staining and fluorescence in situ hybridization for t(14;18) (dual color, break-apart probes) were performed as previously described.25 IHC staining was performed on FFPE tissue sections with the Ventana Benchmark Autostainer (Ventana Medical Systems) using the Ventana 3,3′ diaminobenzidine tetrahydrochloride kit, according to the manufacturer’s instructions. The intensity of BCL2 staining was scored with a semiquantitative scale of staining intensity: intensity similar to or stronger than that of T cells was considered strong, and staining weaker than that of T cells was considered faint. The extent of CD21 follicular dendritic meshwork staining was scored as a percentage of the infiltrate associated with meshwork staining. Meshwork staining associated with <30% of the infiltrate was considered focal staining. Estimation of the proliferation index (PI) score was based on the percentage of Ki67+ B cells within neoplastic infiltrate.
Statistical analysis
Associations between categorical variables were assessed by using Fisher’s exact test, and differences between groups for continuous variables was assessed using a Wilcoxon rank-sum test. Progression-free survival was calculated as the time from diagnosis to the time of relapse or death and was censored at the point when the patient was alive without progression, using the Kaplan-Meier method, and compared by using the log-rank test. P values are 2-sided and considered significant if P < .05.
Results
Clinical features of cutaneous follicular lymphomas
We collected 40 archival skin biopsy specimens of cutaneous FL (Table 1; supplemental Table 1; Figure 1A), including 30 cases of PCFCL and 10 of SCFL. By definition, evaluation for systemic involvement (including computed tomography imaging) was negative in all cases of PCFCL and positive in all cases of SCFL. Of the 30 cases of PCFCL, 26 (87%) remained restricted to skin with no subsequent extracutaneous systemic spread during a median follow-up of 79 months (range, 10-385) (Table 1; Figure 1A). PCFCLs included cases of 13 patients who responded to initial therapy without recurrence and 17 who responded to initial therapy but had recurrence. Strikingly, 13 of 17 recurrences (76%) were restricted to skin at or adjacent to the initial site. In 4 cases of PCFCL, extracutaneous systemic spread (breast, lymph nodes, brain) developed after diagnosis (median, 9 months).
Comparison of clinicopathologic features between cases of PCFCL and SCFL
| PCFCL, n (%) | SCFL, n (%) | P* | |
|---|---|---|---|
| n | 30 | 10 | |
| Sex | |||
| Female | 13 (43) | 5 (50) | .73 |
| Male | 17 (57) | 5 (50) | |
| Age, median (range) | 60 (32-79) | 59 (33-91) | .47 |
| Staging | |||
| IA (cutaneous) | 30 (100) | 0 | <.001 |
| II-IV | 0 (0) | 10 (100) | |
| Tumor Site | |||
| Head and neck | 15 (50) | 6 (60) | .87 |
| Trunk | 12 (40) | 3 (30) | |
| Extremity (leg/arm) | 3 (10) | 1 (10) | |
| Therapy† | |||
| Localized only | 25 (83) | 5 (50) | .09 |
| Systemic | 5 (17) | 5 (50) | |
| Follow-up, mo | 79 | 55 | |
| Recurrence rate | 17/30 (57) | 9/10 (90) | |
| Time to recur, median, mo | 13 | 15 | |
| Recurrence | |||
| Same cutaneous site | 13/17 (76) | N/A | |
| Different cutaneous site | 1/17 (6) | N/A | |
| Extracutaneous Site | 4/17 (24) | N/A | |
| Histology | |||
| Nodular and nodular/diffuse | 22/28 (79) | 8/8 (100) | .30 |
| Diffuse | 6/28 (21) | 0/8 (0) | |
| IHC | |||
| CD10 | |||
| Negative | 6 (29) | 1 (12) | .63 |
| Positive | 15 (71) | 7 (88) | |
| BCL2 | |||
| Negative | 22 (73) | 1 (10) | <.001 |
| Positive | 8 (27) | 9 (90) | |
| CD21 | |||
| <30% | 10 (33) | 0 (0) | .07 |
| ≥30% | 17 (57) | 8 (100) | |
| Ki-67 | |||
| <30% | 6 (25) | 8 (80) | .006 |
| ≥30% | 18 (75) | 2 (20) | |
| Gene rearrangements | |||
| BCL2 | |||
| Negative | 25 (83) | 0 (0) | <.001 |
| Positive | 5 (17) | 9 (100) | |
| BCL6 | |||
| Negative | 27 (96) | 2 (100) | .99 |
| Positive | 1 (4) | 0 (0) |
| PCFCL, n (%) | SCFL, n (%) | P* | |
|---|---|---|---|
| n | 30 | 10 | |
| Sex | |||
| Female | 13 (43) | 5 (50) | .73 |
| Male | 17 (57) | 5 (50) | |
| Age, median (range) | 60 (32-79) | 59 (33-91) | .47 |
| Staging | |||
| IA (cutaneous) | 30 (100) | 0 | <.001 |
| II-IV | 0 (0) | 10 (100) | |
| Tumor Site | |||
| Head and neck | 15 (50) | 6 (60) | .87 |
| Trunk | 12 (40) | 3 (30) | |
| Extremity (leg/arm) | 3 (10) | 1 (10) | |
| Therapy† | |||
| Localized only | 25 (83) | 5 (50) | .09 |
| Systemic | 5 (17) | 5 (50) | |
| Follow-up, mo | 79 | 55 | |
| Recurrence rate | 17/30 (57) | 9/10 (90) | |
| Time to recur, median, mo | 13 | 15 | |
| Recurrence | |||
| Same cutaneous site | 13/17 (76) | N/A | |
| Different cutaneous site | 1/17 (6) | N/A | |
| Extracutaneous Site | 4/17 (24) | N/A | |
| Histology | |||
| Nodular and nodular/diffuse | 22/28 (79) | 8/8 (100) | .30 |
| Diffuse | 6/28 (21) | 0/8 (0) | |
| IHC | |||
| CD10 | |||
| Negative | 6 (29) | 1 (12) | .63 |
| Positive | 15 (71) | 7 (88) | |
| BCL2 | |||
| Negative | 22 (73) | 1 (10) | <.001 |
| Positive | 8 (27) | 9 (90) | |
| CD21 | |||
| <30% | 10 (33) | 0 (0) | .07 |
| ≥30% | 17 (57) | 8 (100) | |
| Ki-67 | |||
| <30% | 6 (25) | 8 (80) | .006 |
| ≥30% | 18 (75) | 2 (20) | |
| Gene rearrangements | |||
| BCL2 | |||
| Negative | 25 (83) | 0 (0) | <.001 |
| Positive | 5 (17) | 9 (100) | |
| BCL6 | |||
| Negative | 27 (96) | 2 (100) | .99 |
| Positive | 1 (4) | 0 (0) |
Statistically significant P values (P < .05) are set in bold. Fisher’s exact test for categorical comparison; Wilcoxon rank-sum test for comparison of age.
Test excluded unknown categories.
Localized therapy: excision, radiotherapy, observation; Systemic therapy: anti-CD20 and chemotherapy.
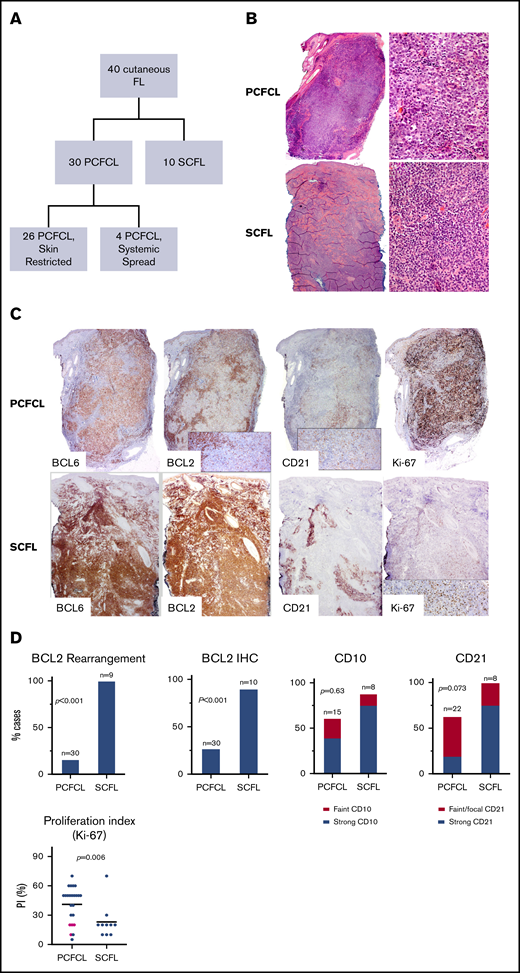
Cutaneous follicular lymphoma cases and their immunohistological characteristics. (A) Summary of the FL cases included in the study. (B) Morphology of cutaneous follicle center lymphomas. PCFCL nodular architecture. Hematoxylin and eosin (H&E) staining, low power (left; original magnification ×40) and high power (right; original magnification ×1000). SCFL. H&E staining, low power (left; original magnification ×40) and high power (right; original magnification ×1000). (C) IHC staining (original magnification ×100) of PCFCL and SCFL specimens: BCL6, BCL2, CD21, and Ki-67. (D) Comparison graphs showing differences in BCL2 rearrangement and BCL2, CD10, CD21, and Ki-67 staining between cases of PCFCL and SCFL. Pink dots in Ki-67 histogram represent cases of PCFCL with systemic spread.
Cutaneous follicular lymphoma cases and their immunohistological characteristics. (A) Summary of the FL cases included in the study. (B) Morphology of cutaneous follicle center lymphomas. PCFCL nodular architecture. Hematoxylin and eosin (H&E) staining, low power (left; original magnification ×40) and high power (right; original magnification ×1000). SCFL. H&E staining, low power (left; original magnification ×40) and high power (right; original magnification ×1000). (C) IHC staining (original magnification ×100) of PCFCL and SCFL specimens: BCL6, BCL2, CD21, and Ki-67. (D) Comparison graphs showing differences in BCL2 rearrangement and BCL2, CD10, CD21, and Ki-67 staining between cases of PCFCL and SCFL. Pink dots in Ki-67 histogram represent cases of PCFCL with systemic spread.
The clinicopathologic features of the cases of PCFCL and SCFL are summarized in Table 1. PCFCL and SCFL had a similar distribution of age and sex. Most PCFCL (27 of 30; 90%) and SCFL (9 of 10; 90%) presented predominantly on the head and neck or trunk. Progression-free survival was longer overall in cases of PCFCL that remained restricted to the skin than that of cases that had subsequent systemic involvement (P = .001; supplemental Figure 1). The median time to recurrence for PCFCL that remained restricted to skin was 24 months (n = 13; range, 5-97), whereas the median time to recurrence of PCFCL with subsequent systemic spread and of SCFL were shorter: 9 months (n = 4; range, 5-36) and 15 months (n = 9; range, 2-73), respectively.
Histopathologic and immunophenotypic characterization of cutaneous follicular lymphomas
The cases of PCFCL had a predominantly dermal infiltrate, often extending into the underlying subcutis, and universally sparing the epidermis (Figure 1B). Most included a predominance of large centrocytes. There was a spectrum of growth patterns, including cases with purely nodular patterns (10 of 28; 36%), nodular and diffuse patterns (12 of 28; 43%), and purely diffuse patterns (6 of 28; 21%). All cases of SCFL had either a purely nodular (3 of 8; 38%) or nodular and diffuse (5 of 8; 63%) pattern (Figure 1B; supplemental Figure 1). All specimens in the cases of SCFL were predominantly composed of centrocytes.
IHC showed that all cases of either type of cutaneous follicular lymphoma were positive for BCL6 (30 of 30 PCFCL and 10 of 10 SCFL). The majority of cases of PCFCL (15 of 21; 71%) and SCFL (7 of 8; 88%) had CD10 expression (P = .63). Although IHC showed that most cases of SCFL strongly expressed BCL2 in the lymphoma cells (9 of 10; 90%), some cases of PCFCL expressed BCL2 (8 of 30; 27%; P < .001; Figure 1C-D; Table 1; supplemental Table 1). Four of the 8 positive cases of PCFCL had subsequent systemic spread.
In contrast to specimens of SCFL that universally showed strong CD21+ follicular dendritic cell meshwork staining (8 of 8; 100%), specimens of PCFCL frequently lacked CD21 staining of follicular dendritic meshwork (10 of 27; 37%; P = .073). Of the PCFCL specimens with CD21+ meshwork staining, the staining was frequently faint (6 of 17) or focal (<30% of infiltrate; 6 of 17). Significantly more cases of PCFCL (18 of 24; 75%) had a high PI (Ki-67 ≥ 30%) than did cases of SCFL (2 of 10; 20%; P = .006; Figure 1D). PCFCL had a median PI of 50% (range, 5%-70%) vs 20% for SCFL (range, 10%-70%). All PCFCL cases that involved systemic spread had a low PI (median 20%; range, 10%-20%).
Assessment of chromosomal rearrangements and copy number alterations in cutaneous follicular lymphomas
In light of the clinical, histologic, and immunophenotypic differences between PCFCL and SCFL, we anticipated that there may be significant genetic differences between these subsets of cutaneous follicular lymphoma. We found that 9 of 9 cases of SCFL but only 5 of 30 cases of PCFL (17%) had a BCL2 rearrangement (P < .001). One case of PCFCL had both BCL2 and BCL6 gene rearrangements (Table 1; supplemental Table 1).
To define copy number alterations and cancer cell fraction, we performed molecular inversion probe array analysis on tumor DNA in 5 cases of PCFCL. Genomic alterations were observed in 4 of 5 cases, including copy number gains, copy number losses, and loss of heterozygosity. The average number of alterations per sample was 11 (range, 1-19) and the average proportion of the genome altered was 18% (range, 5%-44%; supplemental Figure 2). Interestingly, 3 of 5 cases of PCFCL showed loss of chromosome 1p36 (PR08, PS01, and PS02). Other recurrent large genomic alterations observed in at least 2 cases of PCFCL included gains involving chromosomes 7 and 18 and loss of heterozygosity of chromosomes 6p and 9/9p. Recurrent focal copy number alterations included gain of 2p16p15.1 (including REL) and deletions of 2p11.2 (IGKV), 9p21.3 (CDKN2A), and 14q32.33 (IGH locus; supplemental Table 8; supplemental Figure 2). A high copy number gain, defined as >2 copies, was observed in only 1 case of PCFCL (PR01; 2p16.1-p15 region).
Whole-exome sequencing of PCFCL and SCFL tissue specimens
We performed whole-exome sequencing in 18 cases of PCFCL and 6 cases of SCFL. The overall median coverage depth was >150 independent reads per targeted base. Cases of both disease types had a similar mutational burden (median, 5.4 mutations per megabase [range, 1.6-29] and 4.5 mutations per megabase [range, 2.2-13.6], respectively; Figure 2A), comparable to that reported for usual systemic FL.33 C>T transitions, which are associated with UV-induced DNA damage,34 were the most frequent nucleotide substitutions and represented 55% (range, 44%-74%) of variants of PCFCL and 40% (range, 33%-85%) of SCFL (P = .104; Figure 2B). Conversely, C>A transversions, which are rarely UV associated,34 were more commonly seen in SCFL (median, 14%; range, 10%-17%) vs PCFCL (median, 10%; range, 4%-17%; P = .02). There was no difference between the 2 groups in the abundance of activation-induced cytidine deaminase-related mutations (P = .654).
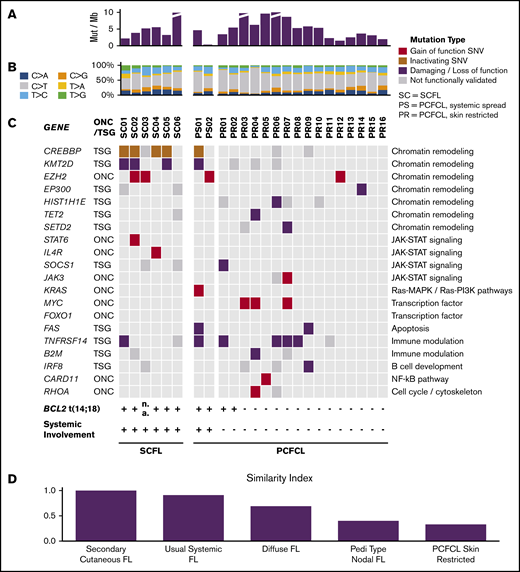
Landscape of somatic alterations in PCFCL and SCFL. (A) The number of somatic mutations per megabase in each specimen. (B) The relative ratios of single somatic nucleotide variant (SNV) types in each specimen. (C) Oncoplot. Red denotes gain of function mutations in putative oncogenes; brown or purple denotes inactivating or damaging mutations in putative tumor-suppressor genes, respectively; gray denotes missense mutations that have not been functionally validated. (D) Similarity index comparison of FL subtypes standardized to SCFL.
Landscape of somatic alterations in PCFCL and SCFL. (A) The number of somatic mutations per megabase in each specimen. (B) The relative ratios of single somatic nucleotide variant (SNV) types in each specimen. (C) Oncoplot. Red denotes gain of function mutations in putative oncogenes; brown or purple denotes inactivating or damaging mutations in putative tumor-suppressor genes, respectively; gray denotes missense mutations that have not been functionally validated. (D) Similarity index comparison of FL subtypes standardized to SCFL.
To maximize our ability to distinguish driver alterations, we used an analytical pipeline designed to identify mutations that have oncogenic or tumor-suppressor signatures in larger disease-relevant data sets. These data sets include systemic FLs,18,20,22,26,35,36 all B-cell lymphomas,37-43 and all cancers (COSMIC).
SCFL.
Using our pipeline, we identified 12 putative cancer-promoting mutations of 6 genes in SCFL (Figure 2C). The majority of these mutations (9 of 12) occurred in genes associated with chromatin remodeling, including 2 EZH2 p.Y646 mutations and 8 loss-of-function or damaging mutations in CREBBP (4 of 6 cases) and KMT2D (3 of 6 cases) (Figures 2 and 3). Three of the 6 cases had mutations in both CREBBP and KMT2D. Known gain-of-function mutations were also identified in STAT6 and IL4R (Figures 2 and 3).
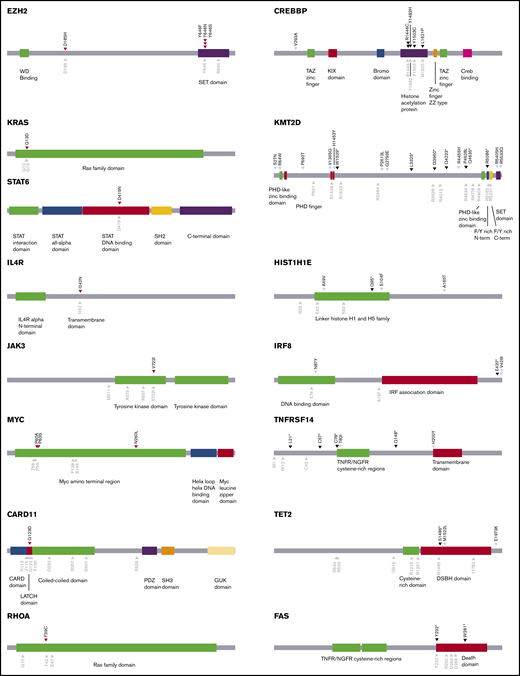
Schematics of genes annotated with oncogenic missense mutations found in CFCL. Mutations marked with red arrowheads (left column) denote validated gain-of-function missense mutations in putative oncogenes. Mutations marked with black arrowheads (right column) denote loss-of-function missense mutations in putative tumor-suppressor genes. Mutations marked with light blue arrowheads (right column) denote missense mutations that have not been functionally validated. Gray arrowheads denote known mutation hotspots.
Schematics of genes annotated with oncogenic missense mutations found in CFCL. Mutations marked with red arrowheads (left column) denote validated gain-of-function missense mutations in putative oncogenes. Mutations marked with black arrowheads (right column) denote loss-of-function missense mutations in putative tumor-suppressor genes. Mutations marked with light blue arrowheads (right column) denote missense mutations that have not been functionally validated. Gray arrowheads denote known mutation hotspots.
PCFCL.
In PCFCL, we identified frequent loss-of-function mutations in TNFRSF14 (5 of 18; 28%) and gain-of-function mutations in MYC (3 of 18; 17%). Interestingly, 2 of the 3 PCFCL samples with 1p36 deletions (PS01, PS02, PR08) also had concomitant damaging TNFRSF14 mutations, which is consistent with a recent report on PCFCL.44 Other validated cancer-promoting mutations included single gain-of-function mutations in JAK3, KRAS, FOXO1, CARD11, and RHOA and loss-of-function mutations in TET2, SOCS1, and B2M (Figures 2 and 3; supplemental Tables 2-7). Two cases had loss-of-function FAS mutations. PCFCL had relatively few mutations in chromatin modifying genes, including 3 of 18 cases with CREBBP and 4 of 18 with KMT2D mutations that have not been functionally validated. Of note, recurrent mutations, including a loss-of-function mutation, were also identified in IRF8 (3 of 18; 17%) and histone H1 genes (9 of 18; 50%). There were 2 cases with SETD2 mutations, a chromatin modifier gene recurrently altered in B-cell leukemias and T-cell lymphomas but not previously described as a recurrently mutated gene in FL.
The PCFCL samples sequenced included 2 samples with subsequent systemic spread. Both cases had t(14;18) translocations. One case had CREBBP and KMT2D inactivating mutations, and the other had an EZH2 p.Y646 mutation.
Comparison of cutaneous follicle center lymphoma and other subtypes of FL
To compare cutaneous B-cell lymphomas with their extracutaneous counterparts, we established a similarity index based on relative mutation prevalence in SCFL or skin-restricted PCFCL of the most commonly mutated genes in each FL subtype.30 Overall, SCFL was genetically more similar to usual systemic FL than the other FL subtypes, and skin-restricted PCFCL was genetically more similar to PTFL than other FL subtypes. Skin-restricted PCFCL and SCFL were the least similar to each other (Figure 2D; supplemental Figures 3 and 4). Key clinical and genetic features of systemic cutaneous FL (SCFL and PCFCL, systemic spread), skin-restricted cutaneous FL (PCFCL, skin-restricted), usual systemic FL, and PTFL are shown in Table 2.
Comparison of distinctive clinicopathologic and genetic features in follicular lymphoma subtypes
| Usual systemic FL % (n/group total)* | Cutaneous FL, systemic % (n/group total)† | Cutaneous FL, skin restricted % (n/group total)‡ | Pediatric type nodal FL % (n/group total)* | |
|---|---|---|---|---|
| BCL2 rearrangement | 89 (16/18) | 92 (12/13) | 8 (2/26) | 0 (0/26) |
| Mutations in 2 or more of 4 chromatin modifiers (CREBBP, KMT2D, EZH2, and EP300) | 63 (47/75) | 63 (5/8) | 6 (1/16) | 4 (1/24) |
| Ki-67 <30% | 67 (10/15) | 86 (12/14) | 10 (2/20) | 0 (0/26) |
| Clinical | ||||
| Multisystemic involvement | Yes, often widely disseminated | Yes, skin+LNs and/or other organs | No, limited to the skin | No, limited to LNs of 1 area |
| Anatomical location | Variable | Head and neck skin predominant | Head and neck skin predominant | Head and neck LN predominant |
| Therapy | Often requires systemic therapy | Often requires systemic therapy | Localized therapy (excision, targeted RT, IL steroids or IL rituximab) | Localized therapy (excision, targeted RT) |
| Presumed site of disease/clone origin | Bone marrow precursor | Bone marrow precursor | Site of anatomically restricted disease | Site of anatomically restricted disease |
| Usual systemic FL % (n/group total)* | Cutaneous FL, systemic % (n/group total)† | Cutaneous FL, skin restricted % (n/group total)‡ | Pediatric type nodal FL % (n/group total)* | |
|---|---|---|---|---|
| BCL2 rearrangement | 89 (16/18) | 92 (12/13) | 8 (2/26) | 0 (0/26) |
| Mutations in 2 or more of 4 chromatin modifiers (CREBBP, KMT2D, EZH2, and EP300) | 63 (47/75) | 63 (5/8) | 6 (1/16) | 4 (1/24) |
| Ki-67 <30% | 67 (10/15) | 86 (12/14) | 10 (2/20) | 0 (0/26) |
| Clinical | ||||
| Multisystemic involvement | Yes, often widely disseminated | Yes, skin+LNs and/or other organs | No, limited to the skin | No, limited to LNs of 1 area |
| Anatomical location | Variable | Head and neck skin predominant | Head and neck skin predominant | Head and neck LN predominant |
| Therapy | Often requires systemic therapy | Often requires systemic therapy | Localized therapy (excision, targeted RT, IL steroids or IL rituximab) | Localized therapy (excision, targeted RT) |
| Presumed site of disease/clone origin | Bone marrow precursor | Bone marrow precursor | Site of anatomically restricted disease | Site of anatomically restricted disease |
IL, intralesional; LN, lymph nodes; RT, radiotherapy.
Cutaneous FL, systemic (n = 14) includes all cutaneous FL with systemic involvement, including cases of SCFL (n = 10) and PCFCL (systemic spread) (n = 4).
Cutaneous FL, skin restricted (n = 26) includes all cutaneous FL that remain restricted to skin, including PCFCL (skin restricted) but not PCFCL (systemic spread) (n = 4). These data are in contrast to Table 1, which includes all cases of PCFCL (n = 30).
Role of immunophenotypic and genetic features in diagnosis and categorization
It is critically important to differentiate cases of cutaneous FL that ultimately disseminate (SCFL and PCFCL with subsequent systemic spread) from those that remain restricted to the skin, given the significant differences in prognosis and therapy. However, making this determination can be challenging, as extracutaneous dissemination is sometimes not detectable at the time of initial diagnosis. There are currently no known tumor-derived biomarkers that can reliably identify cases with concurrent or subsequent systemic involvement at the time of diagnosis.
To this end, we established a novel set of criteria (Figure 4A) based on the relative absence of mutations in chromatin-modifying genes in PCFCL combined with other distinguishing features. We assessed the ability of the following criteria to distinguish PCFCL from SCFL: (1) the presence of mutations in 2 or more of the 4 most commonly mutated chromatin-modifying genes in FL45 (CREBBP, KMT2D, EP300, and EZH2); (2) the presence of BCL2 gene rearrangement; (3) a proliferation fraction <30% (the PI threshold previously established to delineate high proliferation fraction in FL).25,46 Our hypothesis was that the fulfillment of 2 or more of these criteria would predict cutaneous FL cases with systemic spread, and that negativity for 2 or more criteria would predict cases of skin-restricted PCFCL.

Proposed criteria and its application to the study cohort. (A) Proposed criteria for prediction of systemic spread. (B) Application of the criteria to all 40 cases in the study cohort.
Proposed criteria and its application to the study cohort. (A) Proposed criteria for prediction of systemic spread. (B) Application of the criteria to all 40 cases in the study cohort.
Of the 40 cases in the series, 60% (24 of 40) were sequenced, and data were obtained for at least 2 of the 3 criteria in 93% of cases (37 of 40; Figure 4B). Overall, 12 cases were positive for at least 2 of the 3 proposed criteria for predicting systemic involvement. Each of these 12 cases showed systemic involvement, including 9 cases of SCFL and 3 cases initially diagnosed as PCFCL with later systemic spread. Twenty-one other cases were negative for at least 2 of the 3 criteria. Each of these 21 cases were PCFCL that remained localized to skin. The remaining 7 cases did not have sufficient data to apply the proposed criteria. Overall, the application of the criteria resulted in the accurate categorization of all 33 evaluable cases into cutaneous FL with persistent skin restriction (PCFCL) or cutaneous FL with systemic spread (SCFL and PCFCL with later systemic spread) with 100% sensitivity and specificity. This result represents an improvement in accuracy over both the original diagnosis at the time of presentation (sensitivity for systemic disease of 71%) and BCL2 gene rearrangement alone (specificity for systemic disease of 90%; for the 32 cases assessable for both criteria).
Discussion
In this study, we performed, to our knowledge, the largest, most comprehensive genetic analysis to date of cutaneous follicular lymphomas in an effort to characterize the defining genetic features of PCFCL and to identify genetic, immunophenotypic, and histopathologic features that can potentially distinguish cases of skin-restricted PCFCL from cutaneous follicular lymphomas with concurrent (SCFL) or subsequent systemic spread.
Patients with PCFCL or SCFL in our cohort were clinically similar in age, sex, anatomic site of presentation, histology, and most immunophenotypic features (eg, CD10 and BCL6 expression). We confirmed previously reported differences between SCFL and PCFCL, including relative absence of strong BCL2 expression, weaker CD21 follicular dendritic meshwork staining, and higher PI in PCFCL relative to SCFL.
Genetically, we confirmed the rarity of BCL2 rearrangements in PCFCL, in contrast to their near-universal presence in SCFL. We discovered that most of the cases of SCFL had mutations in chromatin-modifying genes (eg, CREBBP, KMT2D, EP300, and EZH2) identical to those previously identified and reported for usual systemic follicular lymphoma, whereas PCFCL lacked these mutations, with the exception of occasional patients with variants of unknown significance. Instead, cases of PCFCL had mutations in other oncogenic pathways, as well as recurrent mutations in IRF8 and/or TNFRSF14. A subset of cases had concurrent deletions of the 1p36 locus that includes TNFRSF14.
Although the ontogeny of follicular lymphoma is poorly understood, usual systemic follicular lymphoma is thought to arise from an early marrow precursor, in which t(14;18) and CREBBP and KMT2D mutations are fundamental, and from transformative genetic alterations that contribute to malignancy (Figure 5A). This notion is supported by the presence of each of these alterations in 80% to 90% of cases of usual systemic FL, including more localized disease, such as duodenal follicular lymphoma.47 The relative absence of these genetic alterations in PCFCL and other FL subtypes, such as PTFL, confirms that these subtypes are distinct biological entities derived from completely different cells of origin than the t(14;18)+ precursor, from which usual nodal systemic FL is derived. Notably, in addition to genetic similarities, PCFCL and PTFL share other clinicopathologic features (eg, high-PI, self-limiting presentation with localized anatomic involvement, and excellent prognosis) that distinguish them from their systemic counterparts (SCFL and usual systemic FL, respectively). This correlation influenced the criteria used for predicting cases with systemic spread (Figure 4A). Although MAPK pathway mutations have been shown to drive PTFL pathogenesis, further studies are needed to identify the PCFCL cell of origin (potentially skin-associated or skin-resident B cells; Figure 5A) and the mechanisms driving its pathogenesis.
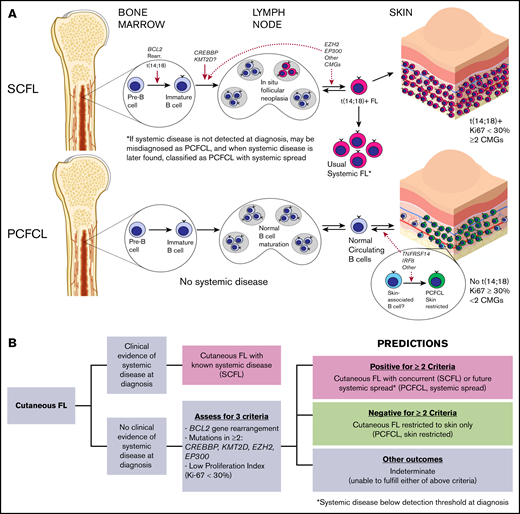
Models of pathogenesis for SCFL and PCFCL and proposed clinical prediction algorithm. (A) Proposed SCFL and PCFCL ontogenies. Solid red arrows denote sites of genetic mutational events supported by the FL literature, and dotted red arrows denote hypothesized sites. CMG, chromatin-modifying genes. (B) Proposed clinical prediction algorithm for approaching newly diagnosed cases of cutaneous follicular lymphoma.
Models of pathogenesis for SCFL and PCFCL and proposed clinical prediction algorithm. (A) Proposed SCFL and PCFCL ontogenies. Solid red arrows denote sites of genetic mutational events supported by the FL literature, and dotted red arrows denote hypothesized sites. CMG, chromatin-modifying genes. (B) Proposed clinical prediction algorithm for approaching newly diagnosed cases of cutaneous follicular lymphoma.
Prior studies assessing BCL2 gene rearrangements and BCL2 expression in cutaneous FL (>300 cases of PCFCL and 50 of SCFL, combined).3,4,6-9,44,48-51 have shown that, although BCL2 gene rearrangements are much more commonly associated with SCFL than PCFCL, a subset of cases of PCFCL (up to 30% of cases) also have this genetic alteration. Almost all cases of SCFL and 20% to 86% of those of PCFCL in prior studies have been shown to have BCL2 expression, including nearly all cases of SCFL and PCFCL that have BCL2 gene rearrangements. All of these studies have demonstrated that despite the stronger association of BCL2 gene rearrangements with SCFL, this genetic alteration is not sufficiently specific to reliably distinguish PCFCL from SCFL or from cases of PCFCL with later systemic spread. Our current study agrees with these findings, with all examined cases of SCFL having BCL2 rearrangements and 90% having BCL2 expression, whereas 17% of cases of PCFCL had BCL2 rearrangements and 27% had BCL2 expression. As part of our genetic analysis, we also found that mutations in chromatin-modifying genes were much more common in SCFL than in PCFCL (similar to BCL2 gene rearrangements), yet were not independently sufficient to distinguish cases of skin-restricted PCFCL from cases of SCFL or PCFCL with subsequent systemic spread.
A 2019 study reported that strong BCL2 expression was the best parameter in that study for identifying SCFL and PCFCL with later systemic spread.3 Although our results generally agree that strong BCL2 expression is associated with SCFL, especially in association with BCL2 rearrangements, we found that BCL2 expression alone is not specific enough to reliably differentiate PCFCL from cutaneous FL with systematic involvement. In addition, BCL2 staining is complicated by the fact that both neoplastic cells and adjacent T cells can express BCL2, and tissue-staining intensity may vary among reagents, tissue types, and laboratories.
To define a reliable and objective approach to distinguishing PCFCL with skin restriction from cutaneous FLs with concurrent or subsequent systemic involvement, we simultaneously assessed the various genetic and immunophenotypic features that tend to differ between PCFCL and SCFL. As part of this approach, we created a novel set of criteria by combining 3 features that distinguish PCFCL and SCFL: (1) the presence of BCL2 rearrangements, (2) a low proliferation fraction (<30%), and (3) mutations in 2 or more of the chromatin-modifying genes CREBBP, KMT2D, EP300, and EZH2. We predicted that positivity for 2 or more of these criteria would predict proclivity for systemic spread, and negativity for 2 or more criteria would predict the likelihood of persistent skin restriction. The thresholds established for each of these 3 criteria were influenced by data from the literature. As already discussed, the rarity of BCL2 gene rearrangements in PCFCL has been well-established, although variably low percentages of cases diagnosed as PCFCL have been shown to have BCL2 gene rearrangements. Similarly, the relatively high PI of PCFCL has been established in a few studies.9,44 The 30% threshold has been established as a cutoff for “high-PI” for FL in several studies.25,46 Finally, given our current discovery that chromatin-modifying gene mutations are not as prominent a feature of PCFCL as they are of SCFL, we chose to include in the criteria the 4 most commonly mutated chromatin-modifying genes in FL.18
Application of this new set of criteria to the cutaneous FL cases in our study not only identified all 9 cases of SCFL with high sensitivity and specificity, but also predicted systemic spread in all 3 cases of cutaneous FL initially diagnosed as PCFCL. It also accurately predicted skin restriction in all 21 additional cutaneous FL cases. Therefore, we propose that these criteria may be used as part of a simple algorithm for predicting the likelihood of systemic spread at the time of initial diagnosis (Figure 5B). As this study represents the first comprehensive genetic study of PCFCL and the first to simultaneously assess BCL2 rearrangements, PI and the mutational profile of cutaneous follicular lymphomas, we cannot fully assess these criteria in prior studies of cutaneous FL. It is certainly important that the utility of these criteria be assessed in future studies of cutaneous follicular lymphoma.
In summary, the findings in this study demonstrate that PCFCL has biologically distinct indolent cutaneous germinal center B cells that lack BCL2 and BCL6 rearrangements and recurrent mutations in chromatin-modifying genes that are often mutated in usual systemic FL. A subset of apparent PCFCLs without extracutaneous involvement at diagnosis that subsequently progress to overt systemic disease have worse prognoses. These cases likely represent rare usual systemic FLs that initially present in the skin, as suggested by their having the genetic alterations seen in usual systemic FL. We propose the application of a simple set of criteria to help distinguish skin-restricted PCFCL from cases of cutaneous FL that are likely to have concurrent or future systemic involvement. This study adds to a growing body of literature that reveals distinctive genetic features supporting the traditional subclassification of primary cutaneous B-cell lymphomas, as defined by histological and clinical criteria.30,39,52 Assessment for their distinctive genetic features may prove helpful in the diagnosis and prognosis of some cases for which the histological distinctions between these subtypes may be challenging.
Original data are available by e-mail request to the corresponding author and by public deposit, if necessary.
Acknowledgments
The authors thank the patients who contributed specimens to this study; the Northwestern Skin Disease Research Center (DNA/RNA Delivery Core), the Robert H. Lurie Comprehensive Cancer Center (Pathology Core), the Center for Cancer Genome Discovery, the Yale Center for Genome Analysis, and Admera Health for their sequencing support; and the Northwestern University Research Computing Services for their invaluable contribution of effective solutions for the storage and analysis of data.
This work was supported in part by the Foglia Family Foundation. A.L. was supported by the National Institutes of Health (NIH), National Cancer Institute (K23CA184279); the American Society of Hematology/Amos Medical Faculty Development Program; and funding from an anonymous donor. J.C. is the Ruth K. Freinkel Research Professor and a Doris Duke-Damon Runyon Clinical Investigator and was supported by NIH, National Cancer Institute (K08-CA191019-01A1); National Center for Advancing Translational Sciences (NCATS) (UL1TR001422); the Damon Runyon Cancer Research Foundation (DRCRF CI-84-16); and the Doris Duke Charitable Foundation (DDCF 2016095). X.Z. was supported by a Dermatology Foundation Medical Dermatology Career Development Award; a Cutaneous Lymphoma Foundation Catalyst Research grant; an institutional grant from the Northwestern University Clinical and Translational Sciences Institute (NUCATS) and NIH, NCATS (KL2TR00142405A1). A.W. was supported by NIH, National Library of Medicine (T15-LM011271).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: X.A.Z., J.Y., K.G.R., A.T.W., D.F., and J.C. performed the genetic analyses; A.L., J.G., and L.C. reviewed pathology and performed associated analysis; M.E.M.-E. and J.G. provided the clinical information and the samples from Northwestern University, and L.C. provided the same for the Austrian samples; H.K.M., A.P.M., and S.H., contributed/curated the MGH cases and contributed to the analysis; E.A.M., contributed/curated the BWH cases and contributed to the analysis; C.N.P., B.H., and E.F.A, provided copy number data and other genetic analyses for the MGH/BWH samples; K.E.S. provided biostatistical analysis and support; D.M.W. contributed to research design and the manuscript; J.C. and A.L. designed the research and oversaw the project; and J.C., A.L., and X.A.Z wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial of interests.
Correspondence: Abner Louissaint Jr, Massachusetts General Hospital, Department of Pathology, 55 Fruit St, Warren 2, Boston, MA 02114, e-mail: alouissaint@partners.org; and Jaehyuk Choi, Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, 303 E Superior St, Room 5-115, Chicago, IL 60611; e-mail: jaehyuk.choi@northwestern.edu.

