

Key Points
BPDCNs have varying profiles evoking either pDC or AS-DC signatures, and phenotype can sometimes be confusing, notably with T-cell ALL.
Genetics is heterogeneous: a lymphoid-like profile (IKZF1) is highly prevalent, whereas immune defects can mimic AS-DC characteristics.

Abstract
Oncogenesis and ontogeny of blastic plasmacytoid dendritic cell neoplasm (BPDCN) remain uncertain, between canonical plasmacytoid dendritic cells (pDCs) and AXL+ SIGLEC6+ DCs (AS-DCs). We compared 12 BPDCN to 164 acute leukemia by Affymetrix HG-U133 Plus 2.0 arrays: BPDCN were closer to B-cell acute lymphoblastic leukemia (ALL), with enrichment in pDC, B-cell signatures, vesicular transport, deubiquitination pathways, and AS-DC signatures, but only in some cases. Importantly, 1 T-cell ALL clustered with BPDCN, with compatible morphology, immunophenotype (cCD3+ sCD3− CD123+ cTCL1+ CD304+), and genetics. Many oncogenetic pathways are deregulated in BPDCN compared with normal pDC, such as cell-cycle kinases, and importantly, the transcription factor SOX4, involved in B ontogeny, pDC ontogeny, and cancer cell invasion. High-throughput sequencing (HaloPlex) showed myeloid mutations (TET2, 62%; ASXL1, 46%; ZRSR2, 31%) associated with lymphoid mutations (IKZF1), whereas single-nucleotide polymorphism (SNP) array (Affymetrix SNP array 6.0) revealed frequent losses (mean: 9 per patient) involving key hematological oncogenes (RB1, IKZF1/2/3, ETV6, NR3C1, CDKN2A/B, TP53) and immune response genes (IFNGR, TGFB, CLEC4C, IFNA cluster). Various markers suggest an AS-DC origin, but not in all patients, and some of these abnormalities are related to the leukemogenesis process, such as the 9p deletion, leading to decreased expression of genes encoding type I interferons. In addition, the AS-DC profile is only found in a subgroup of patients. Overall, the cellular ontogenic origin of BPDCN remains to be characterized, and these results highlight the heterogeneity of BPDCN, with a risk of a diagnostic trap.
Introduction
Dendritic cells (DCs) consist of conventional or myeloid dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs). pDCs are lineage− CD4+ CD123high HLA-DRhigh and produce large amounts of interferon (IFN) type I in response to viral infections.1 They develop from myeloid and/or lymphoid progenitors. The myeloid branch includes common myeloid progenitors (CMPs) and common DC progenitors (CDPs) with both cDC and pDC potential,2,3 while the lymphoid branch involves common lymphoid progenitors.4-6 Recently, AXL+ SIGLEC6+ CD11c−/low DCs (AS-DCs) have been described among the CD123high HLA-DRhigh DCs.4,7 Single-cell RNA sequencing shows that this population shares pDC- and cDC-like enriched signatures. Characterized by low IFN secretion and the ability to stimulate T-cell proliferation, they have properties closer to cDC and might be a transition state from pDC to cDC, explaining conflicting descriptions of pDCs.7,8 They express cDC markers, such as CD2, CD33, CD5, CD86, but also pDC markers, such as CD123high and CD303,7 and present some B-cell features: IGLL1+ SIGLEC1+ CD22+ LYZ+ compared with canonical pDCs.4,7,9-12
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare and aggressive leukemia derived from pDCs, which is characterized by skin involvement, and affects mainly elderly men.13-18 Our team previously described a BPDCN-specific transcriptomic profile leading to dysregulation of LXR target genes involved in cholesterol homeostasis.19 The mutational landscape of BPDCN is close to myeloid malignancies, impacting epigenetics (TET2, ASXL1, EZH2, ATRX, IDH1, IDH2, DNMT3A), the RAS pathway (NRAS, KRAS), splicing (SF3B1, SRSF2, ZRSR2), kinase signaling (FLT3, KIT), and tumor removal (TP53, RB1, ATM).20-27 Moreover, myeloid neoplasms can be associated with or followed by BPDCN,28 especially chronic myelomonocytic leukemia (CMML),29,30 with shared clonal mutations in 2 studies.31,32 Conversely, the rare case reports of BPDCN associated with lymphoid neoplasms never show common genetic events, suggesting cooccurrence instead of a common clonal origin.33-35 Conversely, deletions of CDKN2A/CDKN2B and IKZF1 are particularly prevalent in BPDCN, similarly to lymphoid neoplasms, as well as MYC rearrangements, underpinning similarities with high-grade B-cell lymphoma.25,36-38 Moreover, the CAL-1 cell line, derived from a BPDCN patient, is MYC-rearranged and exhibits an AS-DC profile with the following phenotype: AXL+ SIGLEC1+ IGLL1+ BCL11A+ ID2+ SPIB+ SPI1+ PAX5− TCF3− ID3−.4,37 Variability of lineage marker expression is also frequent in BPDCN, with expression of AS-DC markers, such as CD22, CD2, or CD33.17 However, the exhaustive AS-DC phenotype has never been investigated in BPDCN patients. In addition, some other lymphoid or myeloid lineage markers can be expressed, such as CD7, cCD79a, CD117, and/or CD15, which do not correspond to the AS-DC phenotype.20,18,39-41
Data obtained on BPDCN in recent years have not clarified whether BPDCN derive from canonical pDCs or from AS-DCs, or whether BPDCN can be divided into 2 groups: one derived from canonical pDCs and the other from AS-DCs. In this study, we clarify the transcriptomics signature of BPDCN in comparison with other acute leukemias (ALs) and correlate it with copy number variations (CNV) and mutations to investigate pathways involved in BPDCN oncogenesis and ontogeny.
Methods
Patients and materials
Bone marrow (BM) aspirates or peripheral blood (PB) samples were obtained for BPDCN diagnostic purposes from 12 patients (French BPDCN network, authorization number DC-2008-713) and compared with a control cohort of 164 patients with AL, including acute myeloid leukemia (AML; n = 79), B-cell acute lymphoblastic leukemia (B-ALL; n = 24), and T-cell acute lymphoblastic leukemia (T-ALL; n = 61) (supplemental Methods). All BPDCN patients had cutaneous involvement with diagnostic confirmation on skin biopsies for 10 of them (Table 1). This study was approved by the local ethics committee (CPP EST II, Besançon, France; 31 January 2017).
Clinical and biological features of the cohort
| Patient number | Age, y | Sex | Sample | Infiltration, % | Hematological sites | Extrahematological sites | Prior history (time before diagnosis) | Karyotype | Markers associated to pDC lineage | BPDCN score | Immature lineage markers* | Myeloid lineage markers* | B lineage markers* | T lineage markers* | Anatomopathological confirmation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 38 | M | PB | 65 | PB, BM | Skin | None | 44,X,-Y,del(9)(q11q31),-13[2]/44,idem,add(4q?),add(5q?)[3]/46,XY[14] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | Tdt+ | CD33+ | None | CD2+ | Yes |
| P2 | 69 | M | BM | 90 | BM, LN | Skin | Colon cancer | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303− CD304+ TCL1+ | 3/5 | None | None | None | CD7+ | No |
| P3 | 70 | M | PB | 70 | PB, BM | Skin | CMML | 44,X,-Y,del(6)(q23q25∼27)x2,add(8)(q24),inv(9)(p11q13)c,t(12;15)(p10;p10),-13[18]/46,XY,inv(9)(p11q13)c[2] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ CD117+ | None | Neg | Yes |
| P4 | 51 | M | PB | 71 | PB, BM, LN, spleen | Skin, gums | None | 46,XY,del(6)(q11q22),del(9)(q11q13),del(10)(q12q13) [1]/46,XY,idem,i(7)(q10)[18]/46,XY[1] | CD4+ CD56+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | CD2+ CD5+ low (18%) | Yes |
| P5 | 15 | M | BM | 100 | PB, BM, LN, spleen | Skin, liver | None | 43,XY,t(3;6),der(7)t(7;19),-9,der(12)t(5,12),-13,-19[7]/86,idem x2[3] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | Neg (CD7 and CD2 NA) | Yes |
| P6 | 66 | M | BM | 74 | PB, BM, LN | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | CD22+ | CD7+ | Yes |
| P8 | 60 | M | PB | 70 | PB, BM, LN | Skin | None | 46XY[18]/44, X,-Y, t(4;12),-9,-13,+mar[9] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | None | CD7+ CD2+ | Yes |
| P9 | 74 | M | PB | 75 | PB, BM | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | CD7+ CD2+ | Yes |
| P11 | 59 | M | BM | 95 | PB, BM, LN | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | Neg | Yes |
| P12 | 61 | M | BM | 82 | PB, BM | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303− CD304+ | 3/5 | None | None | None | CD2+ | Yes |
| P18 | 75 | F | PB | 90 | PB, BM, spleen | Skin | None | 45,XX,del(5)(q13q34),?inv(11)(p11q21),der(15)(15qter-15q10::?::18q10- 18qter),-18[15]/44,idem,-22,dmin [2]/88,idem x2,del(6)(q16)[2]/46,XX[1] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ | 5/5 | None | None | None | CD7+ | Yes |
| P89 | 82 | M | PB | 80 | PB, BM, LN | Skin, tonsil | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | None | Neg | No |
| P13 | 46 | F | BM | NA | PB, BM | Absent | None | 45,XX,del(1)(p21),-5,-7, del(11)(q13),del(13)(q13;q22),-15, +mar1[3]/46,XX[17] | CD4+ CD56− CD123+ CD304+ CD303− TCL1+ | 2/5 | Tdt+ | CD117+ | None | cCD3+ sCD3− CD2+ CD5+low (20%) | No |
| Patient number | Age, y | Sex | Sample | Infiltration, % | Hematological sites | Extrahematological sites | Prior history (time before diagnosis) | Karyotype | Markers associated to pDC lineage | BPDCN score | Immature lineage markers* | Myeloid lineage markers* | B lineage markers* | T lineage markers* | Anatomopathological confirmation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 38 | M | PB | 65 | PB, BM | Skin | None | 44,X,-Y,del(9)(q11q31),-13[2]/44,idem,add(4q?),add(5q?)[3]/46,XY[14] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | Tdt+ | CD33+ | None | CD2+ | Yes |
| P2 | 69 | M | BM | 90 | BM, LN | Skin | Colon cancer | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303− CD304+ TCL1+ | 3/5 | None | None | None | CD7+ | No |
| P3 | 70 | M | PB | 70 | PB, BM | Skin | CMML | 44,X,-Y,del(6)(q23q25∼27)x2,add(8)(q24),inv(9)(p11q13)c,t(12;15)(p10;p10),-13[18]/46,XY,inv(9)(p11q13)c[2] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ CD117+ | None | Neg | Yes |
| P4 | 51 | M | PB | 71 | PB, BM, LN, spleen | Skin, gums | None | 46,XY,del(6)(q11q22),del(9)(q11q13),del(10)(q12q13) [1]/46,XY,idem,i(7)(q10)[18]/46,XY[1] | CD4+ CD56+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | CD2+ CD5+ low (18%) | Yes |
| P5 | 15 | M | BM | 100 | PB, BM, LN, spleen | Skin, liver | None | 43,XY,t(3;6),der(7)t(7;19),-9,der(12)t(5,12),-13,-19[7]/86,idem x2[3] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | Neg (CD7 and CD2 NA) | Yes |
| P6 | 66 | M | BM | 74 | PB, BM, LN | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | CD22+ | CD7+ | Yes |
| P8 | 60 | M | PB | 70 | PB, BM, LN | Skin | None | 46XY[18]/44, X,-Y, t(4;12),-9,-13,+mar[9] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | None | CD7+ CD2+ | Yes |
| P9 | 74 | M | PB | 75 | PB, BM | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | CD7+ CD2+ | Yes |
| P11 | 59 | M | BM | 95 | PB, BM, LN | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | None | None | Neg | Yes |
| P12 | 61 | M | BM | 82 | PB, BM | Skin | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303− CD304+ | 3/5 | None | None | None | CD2+ | Yes |
| P18 | 75 | F | PB | 90 | PB, BM, spleen | Skin | None | 45,XX,del(5)(q13q34),?inv(11)(p11q21),der(15)(15qter-15q10::?::18q10- 18qter),-18[15]/44,idem,-22,dmin [2]/88,idem x2,del(6)(q16)[2]/46,XX[1] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ | 5/5 | None | None | None | CD7+ | Yes |
| P89 | 82 | M | PB | 80 | PB, BM, LN | Skin, tonsil | None | 46,XY[20] | CD4+ CD56+ HLA-DR+ CD123+ CD303+ CD304+ TCL1+ | 5/5 | None | CD33+ | None | Neg | No |
| P13 | 46 | F | BM | NA | PB, BM | Absent | None | 45,XX,del(1)(p21),-5,-7, del(11)(q13),del(13)(q13;q22),-15, +mar1[3]/46,XX[17] | CD4+ CD56− CD123+ CD304+ CD303− TCL1+ | 2/5 | Tdt+ | CD117+ | None | cCD3+ sCD3− CD2+ CD5+low (20%) | No |
DOD, dead of disease; F, female; LN, lymph node; M, male; NA, not available; Neg, negative; None, no positive marker.
Only positive markers are indicated. Immature lineage marker: CD34, TdT; myeloid lineage marker: CD33, CD117, CD13, CD15, CD65, CD14, CD64, CD11c, MPO; B lineage marker: CD19, CD20, CD22, CD79a; T lineage marker: cCD3 and sCD3 (cytoplasmic and surface), CD2, CD7, CD4, CD8, TCR αβ, TCR gd; BPDCN score, scoring system for BPDCN diagnostic. 40
Molecular annotations
T-cell receptor (TCR) δ and γ rearrangements and direct sequencing of TCR and NOTCH1/FBXW7 genes were performed as previously described.42 Internal tandem duplications and mutations of the tyrosine kinase domain of FLT3 (FLT-ITD and FLT3-TKD) as well as CEBPα mutations were centrally assessed on ABI 2720 Genetic Analyzer (Thermo Fisher Scientific, Wilmington, NC) with Sequencing Analysis v2.5 software.
NGS and bioinformatics analysis
Next-generation sequencing (NGS) was performed from a HaloPlexHS Target Enrichment System designed for Illumina Sequencing (Agilent) targeting 68 genes (supplemental Table 1) in paired-end, 2 × 150 cycles on a MiSeq platform (Illumina Inc, San Diego, CA). Raw data were analyzed using an in-house bioinformatics pipeline (supplemental Methods).43 Sequencing by the Sanger method was performed on an ABI PRISM 3500 DNA Genetic Analyzer (Thermo Fisher Scientific) with a BigDye Terminator v3.1 Cycle Sequencing kit (Thermo Fisher Scientific).
Single-nucleotide polymorphism array analysis
Genome-wide detection of CNV and copy-neutral loss of heterozygosity were performed using the Genome-Wide Human single-nucleotide polymorphism (SNP) Array 6.0 (Affymetrix, Santa Clara, CA). CNV was identified and depicted with tools listed in the supplemental Methods.
Gene expression profiling and data analysis
Gene expression profiling was obtained using Human Genome U133 Plus 2.0 arrays (Affymetrix) from 176 leukemia samples (12 BPDCN, 164 AL) and compared with 5 normal cell-sorted cDCs and 5 pDCs from the GEO database, GSE28490: GSM705321-25 and 29-33, respectively (see supplemental Methods for analysis and supplemental Table 2 for published signatures).
Statistics
Statistical analyses were performed using Prism software v6 (GraphPad Software, San Diego, CA), using nonparametric tests for quantitative variables with non-Gaussian distribution, Spearman correlation, and the Kruskal-Wallis test, as appropriate. All statistical tests were 2-sided, with 5% significance.
Results
Patients
Eleven men and 1 woman diagnosed with BPDCN (median age 63.5 years old, sex ratio: M/F = 11/1) had cutaneous lesions at diagnosis, and a characteristic phenotype of BPDCN on PB or BM (BPDCN score >3)39 ; with high expression of CD123, HLA-DR and cTCL1; expression of CD4, CD56, CD304, and/or CD303, without expression of CD34, or strong lineage commitment markers, such as MPO, cCD3, cCD79a, CD14, and CD11c. The blastic population also expressed T-cell markers in 7 cases (CD2, CD5, and/or CD7), myeloid markers in 6 patients (CD33 and/or CD117), and 1 case expressed the B-cell marker CD22 (Table 1).
A T-ALL reclassified as BPDCN
By unsupervised analysis, the 12 BPDCN samples all clustered together, close to normal pDCs and cDCs, and apart from other AL. Patient P13, initially diagnosed with T-ALL, also clustered with BPDCN patients (Figure 1A). The diagnosis was thus reconsidered for this 46-year-old woman exhibiting cCD3+ blasts (75%, medium intensity), without expression of surface CD3. According to TCR classification of T-ALL, this case was initially designated as immature IM0 T-ALL, with cTCRβ− and germline TCRδ,ϒ,β. Tumor cells expressed CD4 and CD2, low levels of TdT and CD10, and no CD5 or CD7 expression. Considering our transcriptomic results, pDC markers were evaluated in a second step: despite the absence of expression of CD303 and CD56, CD304 was partially expressed (34%) and CD123 and cTCL1 were expressed at high levels (Table 1; Figure 1B). Morphologically, medullar blasts were characterized by regular nuclei and small nucleoli, no cytoplasmic granulation, small vacuoles, and pseudopodia in some cells (Figure 1C). The BPDCN score was only 2, because of cCD3 positivity, but blasts expressed high levels of CD123, and one of the most specific BPDCN markers, cTCL1. In addition, genetic and cytogenetic aberrations found in this patient were similar to the 12 other BPDCN cases, as described below. Taken together, these results led us to retrospectively classify patient P13 as BPDCN for further analyses.
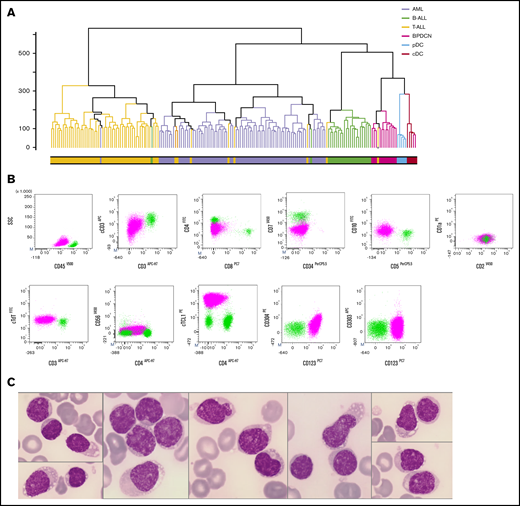
Reclassifying a T-ALL iu BPDCN by gene expression profiling. (A) Unsupervised hierarchical clustering of 176 AL patients and 10 normal DC transcriptomes: B-ALL (green), T-ALL (yellow), AML (lavender), BPDCN (magenta), reclassified patient P13 (yellow with gray dot), normal cDC (red), and normal pDC (light blue). (B) Flow cytometry analysis of patient P13 BM: blastic population (magenta) and CD45+ lymphocytes (light green). The blastic population expresses CD45low cCD3low and CD4+, CD2+, CD10+ but no CD7 and surface CD3. The blastic population expresses the pDC markers CD123, TCL1high and a low level of CD304. CD303 and CD56 were not expressed. (C) BM examination of patient P13 showed blasts, with regular nuclei and small nucleoli, no cytoplasmic granulation, and rare small vacuoles and/or pseudopodia (May-Grünwald-Giemsa stain, magnification ×1000).
Reclassifying a T-ALL iu BPDCN by gene expression profiling. (A) Unsupervised hierarchical clustering of 176 AL patients and 10 normal DC transcriptomes: B-ALL (green), T-ALL (yellow), AML (lavender), BPDCN (magenta), reclassified patient P13 (yellow with gray dot), normal cDC (red), and normal pDC (light blue). (B) Flow cytometry analysis of patient P13 BM: blastic population (magenta) and CD45+ lymphocytes (light green). The blastic population expresses CD45low cCD3low and CD4+, CD2+, CD10+ but no CD7 and surface CD3. The blastic population expresses the pDC markers CD123, TCL1high and a low level of CD304. CD303 and CD56 were not expressed. (C) BM examination of patient P13 showed blasts, with regular nuclei and small nucleoli, no cytoplasmic granulation, and rare small vacuoles and/or pseudopodia (May-Grünwald-Giemsa stain, magnification ×1000).
Transcriptomic signature of BPDCN
A signature of 824 probe sets differentiating BPDCN from other AL was established (supplemental Table 3). Genes associated with pDC lineage were markedly overexpressed in the 13 BPDCN cases, such as LAMP5 (BADLAMP), HES6, LILRB4 (ILT3), LILRA4 (ILT7), IL3RA (CD123), and CLEC4C (CD303), GLUL, IRF7, TLR7, and HLA class II genes, compared with AML, T-ALL, or B-ALL (supplemental Table 3). Some pDC and B-cell markers, such as TCL1A, TCF4, IGLL1, LRMP, and NPC1, displayed expression levels similar to B-ALL in BPDCN, arguing for a B-lymphoid origin (supplemental Figure 1). Furthermore, some AS-DC markers, such as SIGLEC6, were highly expressed in 5 cases, including the CD22+ patient P4 (Figure 2A). Interestingly, another group of 5 cases was isolated (supplemental Figure 2A) using signatures of AS-DCs,7 as well as other signatures: CD5+ CD81+ pDCs,11 pDC commitment,6 activated vs resting pDCs, but also myeloid vs lymphoid lineage26 and hematopoietic progenitors with B-lymphoid potential (hematopoietic stem cells, multilymphoid progenitors and Pro-B).44 This group was not highlighted using signatures defining CMP, granulocyte-monocyte progenitors, megakaryocytic-erythroid progenitors, and earliest thymic progenitor.44 Collectively, gene set enrichment analysis (GSEA) also confirmed that BPDCN were enriched in gene sets upregulated in canonical pDCs as well as AS-DC signatures compared with other leukemias, but also in gene sets involved in protein misfolding, protein transport from the endoplasmic reticulum to the membrane, and inflammatory response to antigenic stimuli (supplemental Table 4).
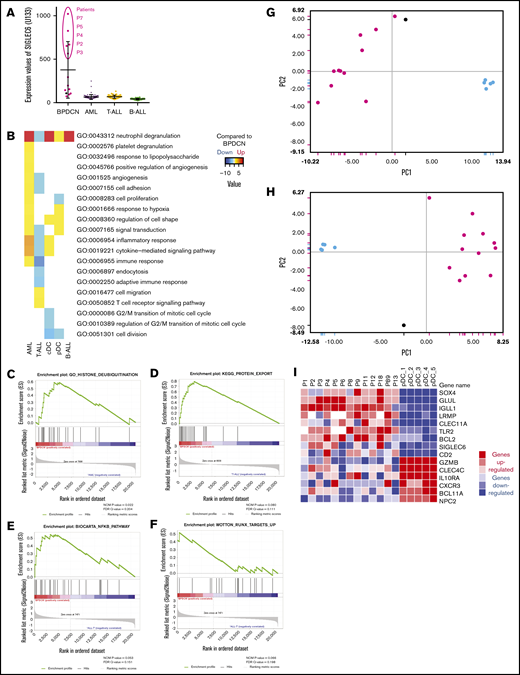
Transcriptomic signature of BPDCN. (A) Expression levels of SIGLEC6 probes on microarray. (B) Main gene ontology pathways deregulated for each type compared with BPDCN, using hypergeometric probability score with 20 HM and P = .05. Downregulated pathways are light blue, and upregulated pathways are yellow to bright red. (C-F) GSEA examples of gene sets enriched in BPDCN compared with AML (C) or T-ALL (D-F). (G-H) Principal component analysis based on published signatures differentiating BPDCN from pDCs (panel G: Sapienza et al26 ; panel H: Villani et al7 ). (I) Heatmap depicting the expression levels of selected upregulated (red) or downregulated (blue) genes in BPDCN compared with normal pDCs. In the dot plot, the 12 BPDCN are depicted in magenta, patient P13 in gray, AML in purple, B-ALL in green, T-ALL in yellow, and normal pDC in light blue.
Transcriptomic signature of BPDCN. (A) Expression levels of SIGLEC6 probes on microarray. (B) Main gene ontology pathways deregulated for each type compared with BPDCN, using hypergeometric probability score with 20 HM and P = .05. Downregulated pathways are light blue, and upregulated pathways are yellow to bright red. (C-F) GSEA examples of gene sets enriched in BPDCN compared with AML (C) or T-ALL (D-F). (G-H) Principal component analysis based on published signatures differentiating BPDCN from pDCs (panel G: Sapienza et al26 ; panel H: Villani et al7 ). (I) Heatmap depicting the expression levels of selected upregulated (red) or downregulated (blue) genes in BPDCN compared with normal pDCs. In the dot plot, the 12 BPDCN are depicted in magenta, patient P13 in gray, AML in purple, B-ALL in green, T-ALL in yellow, and normal pDC in light blue.
Next, we compared BPDCN to each type of AL, beginning with AML. We applied the signature of Dijkman et al,21 which clearly validated the same distinction between BPDCN and AML in our cohort (supplemental Figure 2B). Immune and inflammatory responses and cytokine-mediated signaling pathway appeared upregulated in AML compared with BPDCN, as well as cell adhesion and proliferation (Figure 2B). BPDCN always appeared enriched in pDC gene sets and endoplasmic reticulum genes, but also in the deubiquitination process by GSEA (supplemental Table 5; Figure 2C). When BPDCN were compared with ALL by GSEA, many more pathways were found to be regulated; notably vesicle trafficking pathways and B-cell genes were upregulated in BPDCN, consistent with the B-cell gene enrichment mentioned above (supplemental Table 4; Figure 2D). Interestingly, BPDCN were also enriched in the NF-κB pathway and RUNX targets (Figure 2E-F), corticosteroid pathways, and KMT2A rearrangements by GSEA. Pathways involved in inflammatory or adaptive immune response and in cell organization or signal transduction were downregulated in T-ALL compared with BPDCN (Figure 2B). Finally, when compared with B-ALL, hypergeometric tests did not reveal any significantly different pathway, and GSEA was also less informative (Figure 2B; supplemental Table 4).
The transcriptional signature of BPDCN is clearly different from the pDC signature
As expected, BPDCN transcriptomes were enriched in pDC genes, but differential gene expression analysis revealed a much larger number of microarray probes (1228) that were regulated when we compared the 13 BPDCN with the 5 normal pDC (supplemental Table 3). BPDCN were clearly different from pDCs when applying published signatures (Figure 2G-H).7,26 Genes differentially expressed between normal pDC and BPDCN made it possible to identify oncogenesis pathways in BPDCN. As expected, cell proliferation and division were upregulated in BPDCN, whereas regulation of cell shape and signal transduction was downregulated (Figure 2B).
The top upregulated genes in BPDCN compared with normal pDC included IGLL1, GLUL, CLEC11A, UBE2T, BCL6, TLR2, UBE2C, BCL2, LRMP, UBE2S, and SOX4 (Figure 2I; supplemental Table 3). In contrast, the most relevant downregulated genes in BPDCN compared with normal pDC were GZMB, CLEC4C, NPC1, BCL11A, NPC2, genes regulating the immune system (IL10RA, CXCR3), and ubiquitination (UBE2W, UBE3C) (Figure 2I).
BPDCN exhibit many mutations and chromosomal imbalances
The NGS panel was informative for all cases, with many detected mutations ranging from 1 to 7 (median 3). Twenty genes were found to be mutated among the 68 of the panel (Figure 3A; supplemental Table 5). Epigenetic modifiers were particularly mutated, affecting DNA methylation with TET2 (8/13, 62%), IDH2 (1/13, 8%), or chromatin remodeling with ASXL1 (6/13, 46% including 3 c.1934dupG confirmed by Sanger sequencing), ARID1A (1/13, 8%), and BCOR (1/13, 8%). Other mutations concerned splicing factors ZRSR2 (4/13, 31%), SRSF2 (2/13, 15%); tumor suppressor genes TP53 (2/13, 15%), ATM (2/13, 15%); transcription factors ETV6 (1/13, 8%), IKZF1 (2/13, 15%), IKZF3 (1/13, 8%), ZEB2 (1/13, 8%); RAS pathway NRAS (1/13, 8%), KRAS (1/13, 8%), or cytokine signaling pathway JAK2 (2/13, 15%), STAT3 (1/13, 8%), CXCR4 (1/13, 8%), NOTCH2 (1/13, 8%), and MET (1/13, 8%) (Figure 3A; supplemental Table 5).
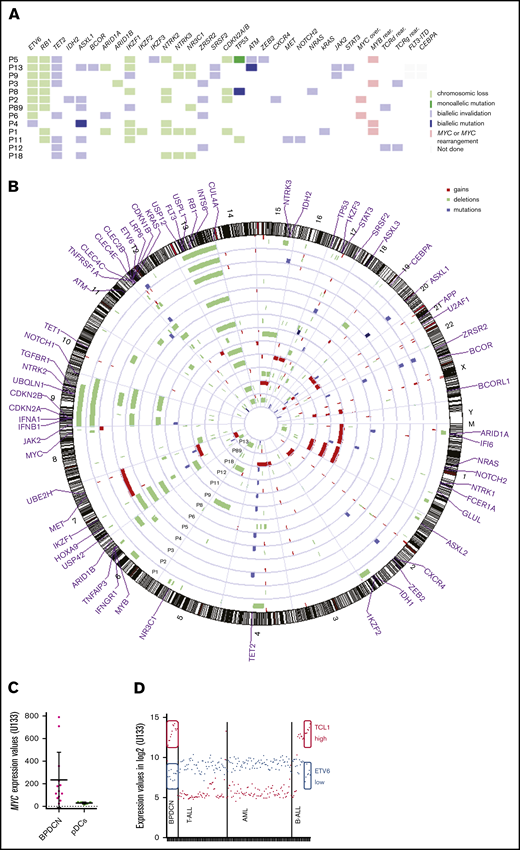
Genetic abnormalities in BPDCN. (A) Mutational profile and key deletions or rearrangements in BPDCN. (B) Circle plot in BPDCN. Blue: biallelic mutation; lavender: monoallelic mutation; lime green: chromosomic loss; green: biallelic invalidation by both chromosome loss and mutation of the other allele; light red: MYC or MYC rearrangement; gray: not done. In the karyogram, deletions are depicted by green bars on the left of the chromosome, gains by red bars on the right of the chromosome, and mutations by blue bars. (C) Expression levels of MYC in BPDCN and pDCs. (D) Expression levels of ETV6 in blue and TCL1A in red on microarrays. High expression of TCL1A and low expression of ETV6 are circled in full line.
Genetic abnormalities in BPDCN. (A) Mutational profile and key deletions or rearrangements in BPDCN. (B) Circle plot in BPDCN. Blue: biallelic mutation; lavender: monoallelic mutation; lime green: chromosomic loss; green: biallelic invalidation by both chromosome loss and mutation of the other allele; light red: MYC or MYC rearrangement; gray: not done. In the karyogram, deletions are depicted by green bars on the left of the chromosome, gains by red bars on the right of the chromosome, and mutations by blue bars. (C) Expression levels of MYC in BPDCN and pDCs. (D) Expression levels of ETV6 in blue and TCL1A in red on microarrays. High expression of TCL1A and low expression of ETV6 are circled in full line.
BPDCN patients also carried many abnormalities in SNP array analysis, with a total of 117 chromosomal imbalances for the 13 BPDCN patients and a median of 9 aberrations per patient (range 2 to 19) (Figure 3B; supplemental Table 6). Deletions (n = 97) were more frequent than gains (n = 20). Patient P13 carried 10 chromosomal losses and 2 chromosomal gains, in agreement with this patient’s requalification as BPDCN.
Notably, the minimal common deletion region (MCDR) on the 7p arm found in 8 patients (61%) included the IKZF1 locus. Moreover, no del(7p) were identified in patients P9 and P89, but stop-gain mutations of IKZF1 (p.E29X and p.Q1429X, respectively) were detected by NGS (Figure 3A; supplemental Table 5). The MCDR 12p13.1-2 included ETV6, CDKN1B, and LRP6 genes (62% of patients). A ninth patient was mutated for ETV6 with a stop-gain p.Q267X (P4), leading to a total of 69% of patients with ETV6 dysregulation. The MCDR 13q14.1-13q14.3, including RB1 and INTS6, was lost in 10 patients (78%), whereas a 9p21.3 MCDR was found in 5 patients (38%), with loss of CDKN2A/CDKN2B and IFN genes. The 9q21.32-21.33 region was also deleted in 6 patients (P1 P2, P4, P5, P8, P18) (46%), notably including the neurotrophic receptor tyrosine kinase NTRK2. The sensitivity of SNP array was assessed by 5q31.3 deletions, including NR3C1, previously investigated for the 12 initially diagnosed BPDCN45 and confirmed here by SNP array. The 15q24.1-15q25.3 MCDR was detected in 4 patients, including NTRK3 (31%). Three patients (P1, P3, P4) had a 6q23.3 deletion (23%), located next to MYB, prompting us to suspect a rearrangement of this gene, known to be involved in recurrent fusion transcript in BPDCN.23 Similarly, a del(8q24) downstream to MYC was detected in patient P2. Expression data were also in favor of an MYC rearrangement, with overexpression of MYC in 2 cases (P2 and P6) compared with other patients (Figure 3C). Two del(17p) were detected, in patients P8 and P11, involving the TP53 locus 17p13.1. One of them (P8) was also TP53-mutated, leading to a biallelic invalidation of the gene (supplemental Table 5). Patient P5 was also mutated for TP53, with a variant allele frequency (VAF) of 98.25%, evoking a loss of heterozygosity, as no deletion was shown. Three patients presented homozygous deletions: in 9p21.3 for patient P1, and in 12p13.2 to p12.3 for patients P2 and P3. A total of 70 genes are included in these deleted regions (supplemental Table 7), including genes of IFN (IFNA1, 2, 4 to 8, 10, 14, 16, 17, 21, IFNB1) and proteins of cholesterol metabolism, such as low-density Lipoprotein Receptor-Related Protein 6 (LRP6).
Impact of chromosomal imbalances on transcriptional expression
When selecting the 354 most relevant genes whose expression is potentially impacted by CNV (supplemental Table 8), ingenuity pathways analysis revealed that the most frequently implicated pathways were infectious disease (24 impaired molecules: 5 gains and 19 losses), cell cycle (41 molecules: 33 loss and 8 gains), developmental disorders (23 molecules: 19 losses and 4 gains), and hematological disease (20 molecules: 14 losses and 6 gains) (supplemental Figure 3; supplemental Tables 8-9). Cellular assembly, organization, and function development were particularly impacted, notably molecular transport, cell-to-cell signaling and cell cycle, as well as genes implicated in immune response, with deletions and downregulation of IFNGR1 (IFN γ receptor 1), TGFBR1, TNFRSF1A (TNF receptor superfamily member 1A), TNFAIP3 (TNF-α-induced protein 3), IFI6 (IFN, α-inducible protein 6), members of the C-type lectin domain family CLEC2B, CLEC4E, and CLEC4C, which were lost in some cases. Cluster A of the HOX family of genes (7p15) and genes of ubiquitination (USP42, UBQLN1, USPL1, CUL4A, USP12) were also recurrently deleted and downregulated, whereas UBE2H was upregulated and gained in 3 patients. Among upregulated genes, GLUL, FCER1A, and APP were also gained in 2 or 3 patients. Moreover, ETV6 was invalidated in most of the patients (9/13), leading to lower expression than in AML and T-ALL. Interestingly, a part of B-ALL expressed ETV6 at the same level as BPDCN, and these B-ALL also highly expressed TCL1A, similarly to BPDCN (Figure 3D). Thus, ETV6 and TCL1A expression was inversely correlated (Spearman, r = −0.2197[−0.3600; −0.0698], P = .0034), and TCL1A is only overexpressed in cases with low ETV6 expression (Kruskal-Wallis, P < .0001).
Discussion
Oncogenesis of BPDCN is still debated, although a growing body of data has accumulated in the last few years.19,23,26,37,45-47 Our study highlights the fact that many oncogenetic processes are activated in BPDCN compared with normal pDCs: cell proliferation and division are upregulated, notably G1/S transition of the mitotic cell cycle. Cell-cycle kinases, such as CDKN1B, CDKN2A, and CDKN2B, are frequently deleted (69%), whereas GLUL, involved in cell proliferation, apoptosis inhibition, and response to glucocorticoids in ALL,48 is upregulated in our cohort of 13 cases compared with pDCs and gained in 3 patients by SNP array. Interestingly, SOX4 is upregulated in BPDCN compared with pDCs. SOX4 is a transcription factor critical for lymphoid and pDC differentiation49 because it upregulates TCF4,50 an E-box transcription factor known to promote pDC lineage commitment,47,51,52 whereas its deletion induces transdifferentiation into cDC cells.53 SOX4 is also expressed in a wide variety of human cancers, promoting migration, invasion, and resistance to apoptosis.54 In hepatocellular carcinoma, cell migration particularly depends of the SOX4 target NRP1, also known as CD304, which is expressed by BPDCN,54 and SOX4 plays a key role in AML leukemogenesis, constituting an independent poor prognostic factor.49,55 Because SOX4 targets TCF4 and NRP1 appear crucial for BPDCN, its upregulation may represent a major element of BPDCN oncogenesis.
Furthermore, 3 cancer-related genes are particularly deleted or mutated: ETV6 (69%), RB1 (69%), and IKZF1 (62%), each of them being crucial for BPDCN.25,31,38,56 In 2 patients, ETV6 deletions were biallelic, evoking an early event, as already suggested,56 and leading to lower expression of ETV6 than in AML and T-ALL. We found that ETV6 and TCL1A expression were inversely correlated and TCL1A is only overexpressed in cases with low ETV6 expression, corresponding to our BPDCN cases and a subgroup of B-ALL, suggesting a link between ETV6 downregulation and TCL1A upregulation. Interestingly, Fears et al57 showed a reverse relationship in B-ALL, with high ETV6 levels associated with decreased endogenous TCL1A expression. Our results highlight an original mechanism of TCL1A deregulation in BPDCN, different from aggressive B-cell lymphoma58,59 and from T-cell prolymphocytic leukemia with chromosomal translocation that makes TCL1A (14q32.13) dependent on TCR enhancer elements.
In addition, NTRK genes were deleted in 9 out of 13 patients (69%) (1 NTRK1, 4 NTRK2, 3 NTRK3, and 1 NTRK2 + NTRK3). Their overexpression and activating mutations are widely described in solid tumors, as well as fusion transcripts, including ETV6-NTRK3 in rare cases of ALL or AML.60 Conversely, loss-of-function mutations are rarer, without a clear role in tumorigenesis.60 Similarly, the impact of such deletions is unknown in BPDCN.
Regulation of cell shape, adhesion, and migration is also impacted in BPDCN. In particular, the HOXA cluster is recurrently deleted, with HOXA9 having an important regulatory role in cytokine induction of endothelium adhesion molecules, ELAM and VCAM-1.61 In addition to the role of the SOX4-NRP1 axis in migration, this deletion could also contribute to BM-to-skin or skin-to-BM cell spreading, as they have protumoral effects, promoting the metastatic process of solid tumors.62-64 HOXA9 and SOX4 deregulation may occur in a second stage, after an early BM oncogenic event, such as the ETV6 deletions, as previously suggested.56
Our study clearly shows high expression of pDC-related genes, consistent with their pDC origin, and confirms activation of the NF-κB and corticosteroid pathways.19,21,26,45 The activation of the canonical NF-kB pathway is particularly interesting, because it would explain the remarkable efficiency of bortezomib in BPDCN, as already demonstrated.26,65 RUNX targets are also enriched in BPDCN, consistent with RUNX2 overexpression, previously described in BPDCN.21 Interestingly, RUNX2 is involved in survival, differentiation, and release of pDCs from BM.66 In BPDCN, its overexpression is considered an oncogenic event, dysregulating pDC differentiation, in some cases through t(6;8)(p21;q24) involving MYC and RUNX2.67
Another key point is the role of pDCs in the immune system, impacted in BPDCN, notably IFN regulation (IFNGR1, IFI6) and pathogen recognition with the CLEC family, including the specific pDC marker CLEC4C (CD303). The latter is known to be negative or low in 25% to 30% of BPDCN cases.17,40
As discussed above, the contribution of the myeloid and lymphoid branch of pDC genesis is still under debate. SIGLEC6, suggestive of the AS-DC cell subtype, appeared upregulated here in 5 cases, as already shown in previous reports in the literature,4,7 and could support an AS-DC origin of these cases, as hypothesized for the CAL-1 cell line.4 However, these results appear sample dependent, as already described, with some primary neoplastic cells able to secrete IFN type I and others not, evoking an AS-DC cell of origin.14 This heterogeneity may suggest that BPDCN could have heterogeneous ontogeny, with some of them being derived from canonical pDCs (7 to 8 cases in our cohort, depending on the signatures used) and others from AS-DCs (5 to 6 cases). As a consequence of the myeloid or lymphoid origin of BPDCN, this heterogeneity could induce variable expression of CD33, CD22, CD2, or CD7 expression, as found in our 13 cases and in our larger cohorts of BPDCN.18,38 However, some AS-DC characteristics could also be explained by the oncogenic process, such as low IFN secretion in BPDCN compared with normal pDCs, which can be attributed to recurrent deletions of IFN genes, or CD2 expression upregulated by SOX4.68 Regrettably, as our cohort would be divided into potential subgroups of <10 cases, we are unable to determine the ontogeny of BPDCN.
The mutational landscape of BPDCN is considered to be myeloid-like (TET2, ASXL1, ZRSR2, SRSF2, BCOR, JAK2), and case reports of CMML evolving to BPDCN30-32 suggest that at least part of BPDCN may originate from a CMP. Nevertheless, the myeloid-like profile of BPDCN cannot be definitively ascertained because genetic data are still limited to small cohorts,22-27 because of the rarity of cases. A lymphoid-like origin of BPDCN is supported by the overexpression of IGLL1, LRMP, BCL11A, BCL2 and genes involved in lymphoid proliferation, such as BCL6, IKZF1, ETV6, and MYC. Of note, IKZF1 abnormalities are highly prevalent in BPDCN, but absent in myeloid neoplasms, except for rare cases in the very particular context of pediatric AML.69 In fact, the myeloid-like profile of BPDCN mainly concerns preleukemic mutations and not transforming events, similarly to AML and MDS arising from clonal hematopoiesis of indeterminate potential (CHIP).70,71 In elderly patients with CHIP, epigenetic abnormalities (TET2, DNMT3A, ASXL1 mutations) typically appear at low VAF (usually <20%), constituting a strong risk factor for subsequent hematological neoplasm (hazard ratio >10).70,71 At the diagnosis of neoplasm, VAF reached higher levels, because of clonal evolution. Similarly, patients are elderly in BPDCN and frequently exhibit TET2 or ASXL1 mutations (85% of our cases). This early epigenetic event could be followed by nonmyeloid oncogenic events, including canonical lymphoid abnormalities (69% of cases with IKZF1/2/3 or CDKN2A/B invalidation, plus ATM, MYC, STAT3) and pan-cancer mutations (NRAS, KRAS, TP53) in the pDC lineage or in cells where the pDC/AS-DC commitment would be promoted by TCF4 upregulation via SOX4 deregulation.
BPDCN displays immunophenotype heterogeneity and misclassification risks. We illustrate this challenge with patient P13. Despite the absence of CD56 and CD303expression, and cytoplasmic expression of CD3, conceptually negative in BPDCN (but already described to be positive intracellularly in BPDCN72 ), a strong commitment to the pDC lineage was shown with gene expression profile, chromosomal imbalances, and positivity of the following markers: CD4, CD123, CD304, and cTCL1. This patient prompts us to perform a BPDCN differential diagnosis in the case of immature AL of uncertain lineage (cCD3+ sCD3− CD7−, without TCRγ and TCRδ rearrangement) when only 1 marker points to T-ALL or AML diagnosis and a fortiori if skin manifestations are detected. Specific markers for differential diagnosis should be used in such circumstances by flow cytometry or immunohistochemistry, such as TCL1, CD123, CD303, RUNX2, HES6, SPIB, and TCF4.21,51,73
In conclusion, the more we learn about pDC ontogeny, the more BPDCN oncogenesis seems complex, with a wide variety of oncogenic mechanisms involved. Developing advanced strategies for diagnosis remains crucial, with mandatory specific markers. We also need to evaluate the clinical and prognostic impact of this heterogeneity in larger cohorts, and the specific deregulated pathways mentioned above should encourage the development of specific targeted therapies in BPDCN. To further investigate potential subgroups with different ontogeny, a validation cohort is required and will be explored by RNA sequencing in an additional 30 cases, coherent with the scarcity of BPDCN.
The data reported in this article have been deposited in the Gene Expression Omnibus (GEO) database, including GSM705329 to GSM705333, and GSE89565. Only transcriptomic data (Affymetrix HG-U133 Plus 2.0 arrays) have been partially presented. In our previous study19: 12 BPDCN out of 13, 65 AML out of 79, and 35 T-ALL out of 61 are available in the GEO database (GSE89565). The 5 primary cDCs and the 5 primary pDCs are also available on the GEO database (GSE28490, GSM705321-25 and 29-33, respectively).
Acknowledgments
The authors thank Fiona Ecarnot for English proofreading. They thank the Laboratory of Immunology (E. Seilles), the Laboratory of Cytology (F. Schillinger), the French BPDCN network, the Groupe Français d’Hématologie Cellulaire (GFHC), the Groupe d’Étude Immunologique des Leucémies (GEIL), the Groupe Français de Cytogénétique Hématologique (GFCH), and the Société Française d’Hématologie (SFH).
This work was supported by La Ligue Contre le Cancer 2008, Fondation ARC (Aides Individuelles DOC20170505805), Institut National de Cancer Translational Research program (DM/FC/sl/RT07), and Association Laurette Fugain (ALF 2018/08).
Authorship
Contribution: F.G.-O., C.R., E.M., and V.A. conceptualized the project; F.G.-O., E.M., and C.R. supervised the research; C.R., A.R., F.R., L.S., S. Biichle, F.A.-D., S.G., and C.F. performed experiments; F.R., M. Cheok, C.R., A.G., P.-J.V., F.J. performed bioinformatic analyses; C.R., F.G.-O., E.M., V.A., C.P. were responsible for patient samples; V.H., S. Bouyer, V.S., P. Saussoy, J.F., and P.F. contributed to the collection of samples, and clinical and biological data for the French BPDCN network; T.P. provided help with immunohistochemistry and anatomopathology analysis; F.R., A.R., F.G.-O., and C.R. wrote the manuscript; C.P., E.M., V.A., P. Saas, C.F., E. Deconinck, E. Daguindau, O.A., M. Callanan, F.J., and J.C. commented on the manuscript; and all authors provided input, edited, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Francine Garnache-Ottou, UMR1098, Laboratoire Hématologie, EFS/B-FC, 8 Rue Dr JFX Girod, 25020 Besançon, France; e-mail: francine.garnache@efs.sante.fr.

