

TO THE EDITOR:
It was with great enthusiasm that we read the recently published work by Yang and colleagues entitled “13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking.”1 In their very well-conducted study, the authors show a novel mechanism of FLT3 disruption in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). In 2019, we had anticipated that unknown mechanisms were to be discovered in acute leukemias with FLT3 overexpression.2 With this in mind, we highlighted that a substantial proportion of either BCP-ALL, T-cell acute lymphoblastic leukemia (T-ALL), or acute myeloid leukemia (AML) cases with FLT3 overexpression lacked a known mechanism leading to this upregulation. Yang et al described in their article that somatic 13q12.2 deletions were present in approximately 2% of all BCP-ALL cases included in the study (5 different cohorts have been evaluated), and they discovered that these deletions lead to high expression levels of FLT3 through chromatin remodeling and enhancer hijacking. In brief, the 13q12.2 microdeletion results in cis interactions between the FLT3 promoter and an enhancer element in intron 8 of PAN3, which ultimately leads to FLT3 upregulation. Although a population-based cohort study is still needed to define the actual frequency of these deletions in BCP-ALL, it is important to note that Yang and colleagues also reported that the 13q12.2 deletions are more frequent in high hyperdiploid BCP-ALL cases. However, other acute leukemia subtypes have not yet been evaluated.
Dr. Spencer has wisely commented on Yang and colleagues’ work, adding that one of the reasons why their findings is significant is that detection of 13q12.2 deletions could have clinical implications for BCP-ALL patients because FLT3 inhibitors are available for targeted therapy. Based on this, Dr. Spencer highlighted that it would be important to investigate whether 13q12.2 deletions also occur in other acute leukemia subtypes in which FLT3 gene alterations are relevant, such as AML.3 In the meantime, we sought to check if this novel mechanism could also explain the FLT3 overexpression recurrently observed in a fraction of T-ALL and AML patients.
Taking advantage of the genomic data sets available, we analyzed a total of 264 T-ALL and 292 AML samples from the Therapeutically Applicable Research to Generate Effective Treatments program (TARGET) ALL phase 2 cohort and TARGET AML cohort, respectively (Figure 1A). The T-ALL samples consisted of 264 primary samples and the AML samples included 102 primary and 95 matched primary and recurrent samples. The T-ALL samples were selected based on the availability of public data to evaluate copy number variations (CNVs) by Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix), which has been analyzed according to the intern pipeline developed at St. Jude Children’s Research Hospital. In summary, to generate copy number data, Affymetrix intensity (.CEL) and the single-nucleotide protein call files have been analyzed using the birdseed (version 2) algorithm into dChip, and probe-level values were summarized.4 After data normalization using the reference normalization algorithm,5 the paired circular binary segmentation has been applied6 with thresholds set to detect copy number segments >2.3 or <1.7 copies covering at least 8 probes (https://ocg.cancer.gov/programs/target/target-methods). Thus, based on these preanalyzed data, we evaluated the presence of 13q12.2 deletions in T-ALL samples. The AML patients were selected based on the availability of whole genome sequencing data that have been analyzed according to the Complete Genomics Assembly Pipeline (version 1.12). In this case, the determination of somatic CNVs was carried out through a coverage sequence computation followed by the estimation and correction of the guanine-cytosine (GC) content bias in coverage of the tumor and matched normal samples. Then, each tumor GC-corrected coverage estimation was normalized by comparison with matched normal GC-corrected coverage. After normalization, the ploidy inference, segmentation, and scoring of samples were performed using hidden Markov models (for more details: https://www.completegenomics.com/documents/CNV+Methods.pdf). These preanalyzed data were deposited in a specific file for each AML patient containing the estimated ploidy for each genome segment. Although the microarray CNV analysis allows the detection of alterations ranging from 40 bp to 8 Mbp,7 the whole genome sequencing makes it possible to detect all types of genomic variation, especially small deletions and duplications, even smaller than 1 kbp.8,9 Once the 13q12.2 microdeletions found by Yang et al varied from 39 to 160 kbp (median size, 129 kbp), we were able to accurately assess the copy number status within the 13q12.2 region in our study cohort.
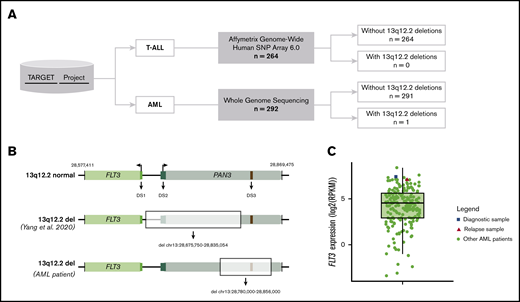
Study overview. (A) TARGET data used for copy number variants assessment in the 13q12.2 region in either T-ALL or AML patients. (B) Schematic figure of the region where FLT3 (light green rectangle) and PAN3 (light blue rectangle) genes are located. (Upper) Wild-type scenario. (Middle) Largest 13p12.2 deletion found by Yang et al. (Lower) Location of 13q12.2 deletion found in the AML patient (highlighted rectangle). DS1 (FLT3 promoter, chr13:28 674,500-28 675,000; dark green rectangle), DS2 (chr13:28 710,000-28 715,000; dark blue rectangle) and DS3 (PAN3 intron 8; chr13:28 843,000-28 843,500; red rectangle). (C) Dispersion of FLT3 expression among AML patients demonstrated by boxplot. The dot plot shows the FLT3 expression of each AML patient (green circle), highlighting the diagnostic (blue square) and relapse samples (red triangle) from the female patient who presented a 13q12.2 deletion. These expression data were obtained according to the availability of RNA-sequencing data from patients assessed for copy-number abnormality (n = 203).
Study overview. (A) TARGET data used for copy number variants assessment in the 13q12.2 region in either T-ALL or AML patients. (B) Schematic figure of the region where FLT3 (light green rectangle) and PAN3 (light blue rectangle) genes are located. (Upper) Wild-type scenario. (Middle) Largest 13p12.2 deletion found by Yang et al. (Lower) Location of 13q12.2 deletion found in the AML patient (highlighted rectangle). DS1 (FLT3 promoter, chr13:28 674,500-28 675,000; dark green rectangle), DS2 (chr13:28 710,000-28 715,000; dark blue rectangle) and DS3 (PAN3 intron 8; chr13:28 843,000-28 843,500; red rectangle). (C) Dispersion of FLT3 expression among AML patients demonstrated by boxplot. The dot plot shows the FLT3 expression of each AML patient (green circle), highlighting the diagnostic (blue square) and relapse samples (red triangle) from the female patient who presented a 13q12.2 deletion. These expression data were obtained according to the availability of RNA-sequencing data from patients assessed for copy-number abnormality (n = 203).
Based on the CNV analysis for T-ALL samples, the specific 13q12.2 deletion has not been found in any patient. Among 292 AML samples, only 1 patient was identified with a 13q12.2 deletion of 76 kb (chr13:28 780,000-28 856,000). The leukemic sample presented a remarkably high level of FLT3 expression (∼3.6 times greater than the mean). This sample belongs to a female patient who was first diagnosed at 4 months old and treated according to the AAML0531 protocol. Unfortunately, she relapsed at 8 months old and died about 1 year after. However, unlike what has been described by Yang et al, we observed that the deletion found in this AML case affects exclusively the PAN3 gene region, including the enhancer element in intron 8 (Figure 1B). It is remarkable that this alteration was identified in the recurrent but not in the primary sample of this case, especially because Yang et al showed that the 13q12.2 deletions they described were highly enriched in cases that subsequently relapsed. Besides, they also identified 1 BCP-ALL case harboring the 13q12.2 deletion at relapse but not at diagnosis, suggesting a later occurrence. However, we cannot exclude the possibility that this PAN3 deletion is present in a small percentage of cells in the diagnostic sample. Nonetheless, by analyzing expression data in these 2 time points, we noticed that there was no difference in FLT3 messenger RNA expression levels between both samples, indicating that this deletion is probably not the mechanism behind FLT3 overexpression in this patient (Figure 1C). Moreover, there was no evidence of allele-specific expression for the FLT3 gene by analyzing the aligned RNA-sequencing data. It is important to mention that none of the samples (diagnostic and relapsed) had any FLT3 amplification or known activating mutations. On the other hand, both had a t(4;11)(q21;q23) translocation, which results in the KMT2A-AFF1 (or MLL-AF4) fusion gene. The presence of this gene rearrangement is known to be directly associated with FLT3 overexpression.10 It is important to note that this patient was diagnosed with the AML-M5 subtype according to the French-American-British classification, which has also been associated with FLT3 overexpression.11,12
The most common alteration associated with FLT3 overexpression is FLT3 activating mutations, mainly in AML,2,13,14 and in some ALL subtypes such as high hyperdiploid BCP-ALL, BCR-ABL1-like (Ph-like) BCP-ALL, and “early T-cell precursor” ALL.15 However, other genetic alterations have also been associated with FLT3 overexpression. These emerging mechanisms of FLT3 transcriptional regulation are summarized in Table 1. In some cases, these alterations are crucial for leukemogenesis and, consequently, they constitute relevant biomarkers for those leukemias. These examples demonstrate the complexity of elucidating the mechanisms underlying FLT3 regulation, especially when dealing with varied disease contexts.
Mechanisms of FLT3 transcriptional deregulation in acute leukemias
| Subtype | Experimental model | Genetic alteration | Mechanism | References |
|---|---|---|---|---|
| B-cell precursor acute lymphoblastic leukemia (BCP-ALL) | BCP-ALL patient samples (n = 1418); GM12878 and NALM-6 cell lines | 13q12.2 deletion | 13q12.2 deletions disrupt chromatin structure and change promoter-enhancer interactions in this region (enhancer hijacking), leading to increased expression of the FLT3 gene | 1 |
| Pro-B-cell lines; bone marrow progenitors; Pax5−/− mice; and case report | PAX5 deletion | It was already demonstrated that Pax5 represses Flt3 transcription in B-cell progenitors, and that Pax5-deficient pro-B cells express abundant Flt3 levels. Based on that, it has been suggested that PAX5 heterozygous deletion in B-cell acute leukemia could be related to FLT3 overexpression | 16,17 | |
| T-cell acute lymphoblastic leukemia (T-ALL) | T-ALL patient samples (n = 60); human leukemia xenograft model; and Jurkat cell line | Loss-of-function alterations of SUZ12, EED, and EZH2 | PRC2 inactivation, due to loss-of-function alterations of SUZ12, EDD, and EZH2, leads to the loss of H3K27me3 in FLT3 promoter region resulting in increased FLT3 expression | 18 |
| Acute myeloid leukemia (AML) | AML patients (TCGA, n = 158); and leukemia stem cells from AML mouse model | DNMT1 haploinsufficient | DNMT1 haploinsufficiency may lead to hypomethylation of FLT3 promoter region, which results in its increased expression | 14 |
| MV4-11, K562, and THP1 human leukemia cell lines | Loss of MSI2 | FLT3 expression can also be regulated by a post-transcriptional mechanism through the MSI2 protein, which physically binds to FLT3 messenger RNA transcripts. In case of genetic loss of MSI2, negative regulation of FLT3 expression is observed, and impaired leukemic growth | 19 | |
| MV4-11 and THP-1 cell lines; THP-1 xenograft murine model | Inhibition of PRMT5 | The PRMT5-Sp1 transcription repressor complex is capable of silencing miR-29b via dimethylation of histone 4 arginine residue H4R3. As a result, an increase in Sp1 is observed, as it is a bona fide target of miR-29b. This event in turn leads to the activation of FLT3 transcription. Thus, the inhibition of PRMT5 can result in a significant increase in the expression of miR-29b and consequent suppression of Sp1 and FLT3 | 20 | |
| Murine-cultured model and bone marrow samples from 104 AML patients | CEBPA biallelic mutations | Together, HOXA9, MEIS1, MYB, and C/EBPα are important elements in FLT3 regulation. In this way, it was observed that CEBPA bi-allelic mutations, mainly in the absence of FLT3 activating mutations, are associated with reduced levels of FLT3 transcript | 21 |
| Subtype | Experimental model | Genetic alteration | Mechanism | References |
|---|---|---|---|---|
| B-cell precursor acute lymphoblastic leukemia (BCP-ALL) | BCP-ALL patient samples (n = 1418); GM12878 and NALM-6 cell lines | 13q12.2 deletion | 13q12.2 deletions disrupt chromatin structure and change promoter-enhancer interactions in this region (enhancer hijacking), leading to increased expression of the FLT3 gene | 1 |
| Pro-B-cell lines; bone marrow progenitors; Pax5−/− mice; and case report | PAX5 deletion | It was already demonstrated that Pax5 represses Flt3 transcription in B-cell progenitors, and that Pax5-deficient pro-B cells express abundant Flt3 levels. Based on that, it has been suggested that PAX5 heterozygous deletion in B-cell acute leukemia could be related to FLT3 overexpression | 16,17 | |
| T-cell acute lymphoblastic leukemia (T-ALL) | T-ALL patient samples (n = 60); human leukemia xenograft model; and Jurkat cell line | Loss-of-function alterations of SUZ12, EED, and EZH2 | PRC2 inactivation, due to loss-of-function alterations of SUZ12, EDD, and EZH2, leads to the loss of H3K27me3 in FLT3 promoter region resulting in increased FLT3 expression | 18 |
| Acute myeloid leukemia (AML) | AML patients (TCGA, n = 158); and leukemia stem cells from AML mouse model | DNMT1 haploinsufficient | DNMT1 haploinsufficiency may lead to hypomethylation of FLT3 promoter region, which results in its increased expression | 14 |
| MV4-11, K562, and THP1 human leukemia cell lines | Loss of MSI2 | FLT3 expression can also be regulated by a post-transcriptional mechanism through the MSI2 protein, which physically binds to FLT3 messenger RNA transcripts. In case of genetic loss of MSI2, negative regulation of FLT3 expression is observed, and impaired leukemic growth | 19 | |
| MV4-11 and THP-1 cell lines; THP-1 xenograft murine model | Inhibition of PRMT5 | The PRMT5-Sp1 transcription repressor complex is capable of silencing miR-29b via dimethylation of histone 4 arginine residue H4R3. As a result, an increase in Sp1 is observed, as it is a bona fide target of miR-29b. This event in turn leads to the activation of FLT3 transcription. Thus, the inhibition of PRMT5 can result in a significant increase in the expression of miR-29b and consequent suppression of Sp1 and FLT3 | 20 | |
| Murine-cultured model and bone marrow samples from 104 AML patients | CEBPA biallelic mutations | Together, HOXA9, MEIS1, MYB, and C/EBPα are important elements in FLT3 regulation. In this way, it was observed that CEBPA bi-allelic mutations, mainly in the absence of FLT3 activating mutations, are associated with reduced levels of FLT3 transcript | 21 |
In conclusion, our work has led us to conclude that 13q12.2 deletion is not a recurrent event in T-ALL nor AML cases. Therefore, our work adds novel and important data to Yang and colleagues’ findings, indicating that this newly described mechanism is specifically associated with FLT3 overexpression in BCP-ALL cases. Moreover, once we excluded the possibility that this mechanism plays a role in T-ALL or AML, an opportunity remains open for future studies that aim to elucidate the mechanisms behind FLT3 overexpression in those leukemia subtypes.
Acknowledgments:
The results published here are in whole based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) initiative, phs000218. The data used for this analysis are available at https://portal.gdc.cancer.gov/projects.
The authors thank the Bioinformatics Core Facility (INCA-RJ) for their support.
Contribution: C.P.P. performed data analyses, interpreted the results, and wrote the manuscript; M.B. and M.E. designed the study, supervised all analyses, and wrote the manuscript; and all authors critically reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mariana Emerenciano, Division of Clinical Research, Research Center–Instituto Nacional de Câncer, Rua André Cavalcanti, 37, 3° floor, Rio de Janeiro, RJ 20231-050, Brazil; e-mail: memerenciano@inca.gov.br.

