

Key Points
Mesothelin is aberrantly expressed in over one third of childhood and young adult AML and not expressed on normal hematopoietic cells.
Mesothelin is successfully targeted in vitro and in xenograft models of MSLN+ AML with ADCs.

Abstract
In an effort to identify acute myeloid leukemia (AML)-restricted targets for therapeutic development in AML, we analyzed the transcriptomes of 2051 children and young adults with AML and compared the expression profile with normal marrow specimens. This analysis identified a large cohort of AML-restricted genes with high expression in AML, but low to no expression in normal hematopoiesis. Mesothelin (MSLN), a known therapeutic target in solid tumors, was shown to be highly overexpressed in 36% of the AML cohort (range, 5-1077.6 transcripts per million [TPM]) and virtually absent in normal marrow (range, 0.1-10.7 TPM). We verified MSLN transcript expression by quantitative reverse transcription polymerase chain reaction, confirmed cell surface protein expression on leukemic blasts by multidimensional flow cytometry, and demonstrated that MSLN expression was associated with promoter hypomethylation. MSLN was highly expressed in patients with KMT2A rearrangements (P < .001), core-binding factor fusions [inv(16)/t(16;16), P < .001; t(8;21), P < .001], and extramedullary disease (P = .001). We also demonstrated the presence of soluble MSLN in diagnostic serum specimens using an MSLN-directed enzyme-linked immunosorbent assay. In vitro and in vivo preclinical efficacy of the MSLN-directed antibody-drug conjugates (ADCs) anetumab ravtansine and anti-MSLN–DGN462 were evaluated in MSLN+ leukemia cell lines in vitro and in vivo, as well as in patient-derived xenografts. Treatment with ADCs resulted in potent target-dependent cytotoxicity in MSLN+ AML. In this study, we demonstrate that MSLN is expressed in a significant proportion of patients with AML and holds significant promise as a diagnostic and therapeutic target in AML, and that MSLN-directed therapeutic strategies, including ADCs, warrant further clinical investigation.
Introduction
Curative treatment of acute myeloid leukemia (AML) remains challenging, despite intensive cytotoxic chemotherapy and hematopoietic stem cell transplant.1 Targeted and immunotherapeutic strategies, including antibody-drug conjugates (ADCs) and adoptive cellular therapies, hold great promise as potent targeted treatments to improve survival and reduce treatment-related toxicity. Utilization of these therapeutic strategies in AML is in its infancy and has been limited by shared expression of many potential target surface antigens on hematopoietic cells.
Large-scale discovery-phase next-generation sequencing efforts in AML, including the collaborative National Cancer Institute/Children’s Oncology Group (COG) Therapeutically Applicable Research to Generate Effective Treatments (TARGET) AML Initiative and The Cancer Genome Atlas (TCGA), evaluating childhood and adult AML patient cohorts, respectively, fueled the identification of target candidates.2-5 Building on the transcriptome analysis, we identified mesothelin (MSLN) to be a highly and uniquely expressed cell surface protein. MSLN is well known as a cell surface marker in numerous solid tumors, including mesothelioma and ovarian, colorectal, and pancreatic adenocarcinomas; it is widely considered a significant potential therapeutic target and is undergoing evaluation in clinical trials.6-9
In nondiseased states, MSLN is expressed in a circumscribed set of tissues, including mesothelial cells lining the pleura, pericardium, and peritoneum. MSLN’s role is unknown, but it is hypothesized to be involved in cell adhesion in healthy and malignant cells. This is supported by the association of MSLN overexpression with increased metastases in various MSLN-overexpressing (MSLN+) tumors.10-12 Notably, MSLN-knockout mice exhibit normal growth, reproduction, and blood counts.13 Elevated surface expression in tumors and apparent dispensable function in normal tissues make MSLN an attractive potential therapeutic target, with a variety of agents (ADCs, immunotoxins, vaccines, and chimeric antigen receptor T cells) currently in development. Clinical trials of MSLN-targeted agents have not demonstrated toxicities attributable to on-target/off-tumor effects.14-18
MSLN has attracted attention as a potential disease marker in its membrane-bound and soluble forms. The precursor product of MSLN anchored at the cell surface undergoes posttranslational modifications, including protease cleavage, yielding 3 main products: (1) cell surface MSLN, the glycosylphosphatidylinositol-anchored N-terminal portion, (2) soluble MSLN, released from glycosylphosphatidylinositol into the extracellular space, and (3) megakaryocyte-potentiating factor (MPF), the soluble C-terminal portion that has no clear function.19,20 Patients with MSLN expression detected on solid tumors often have elevated blood levels of soluble MSLN. Mesomark, an enzyme-linked immunosorbent assay (ELISA)–based blood test, utilizes serum MSLN as a diagnostic marker and is approved by the US Food and Drug Administration for mesothelioma diagnosis and monitoring.21,22
In this study, we describe MSLN as a novel cell surface marker in AML across the age spectrum and the clinical characteristics associated with MSLN overexpression, as well as demonstrate successful therapeutic targeting with MSLN-targeted ADCs in vitro and in vivo.
Methods
Patients
Diagnostic samples were collected from 2051 pediatric patients with de novo AML (ages 1 week to 29.59 years) who were enrolled in COG trials (supplemental Methods), the details of which were described previously.1,23-25 Clinical karyotyping and polymerase chain reaction (PCR)–based molecular testing for NPM1, WT1, FLT3-internal tandem duplication (ITD), and CEBPα were available for 2007 patients (95%). Paired diagnostic relapse samples were available for 263 patients. RNA sequencing (RNA-Seq) data for adult de novo AML patients was obtained from the TCGA database (n = 200, age, 18.2-88.5 years) and the BEAT AML trial (n = 210; ages 21-85 years; supplemental Methods).26,27 Diagnostic samples from 43 adult patients with de novo AML (age, 18-59 years) were obtained in collaboration with the MD Anderson Cancer Center (MDACC; supplemental Methods). The institutional review boards of all participating institutions approved the clinical and research protocols. The study was conducted in accordance with the Declaration of Helsinki.
Genomic characterization
Transcriptome sequencing (RNA-Seq) was performed on diagnostic bone marrow (n = 1411) or peripheral blood (PB; n = 260) from 1671 AML patients, as well as on normal bone marrow (NBM; n = 69) controls and CD34+ cells (n = 17) collected from PB following granulocyte colony-stimulating factor stimulation, as previously described.2 Gene expression is expressed in transcripts per million (TPM). Within this cohort, subsets were further analyzed by targeted capture sequencing (n = 529), microRNA (miRNA) sequencing (n = 1227) with an Illumina Hi-Seq 2000, and DNA methylation analysis (n = 525) with an Infinium HumanMethylation27 or HumanMethylation450 BeadChip Kit (Illumina), as previously described.2
Quantitative reverse transcription PCR
MSLN transcript levels were quantified by quantitative reverse transcription PCR (qRT-PCR) from diagnostic samples for 619 pediatric AML, 41 adult AML (MDACC), and 16 NBM specimens (supplemental Methods).
Sandwich ELISA for soluble MSLN and MPF
Soluble MSLN and MPF were measured in serum collected at diagnosis from 336 pediatric and 43 adult AML patients (at MDACC) by sandwich ELISA, using a Human Mesothelin ELISA MAX Deluxe Kit (BioLegend).
Flow cytometry for cell surface MSLN
Multidimensional flow cytometry (MDF) was performed on diagnostic samples from 158 pediatric and 43 adult AML patients, and MSLN expression using mean fluorescence intensity (MFI) of the myeloid progenitor population was determined using previously described methods.28 The anti-MSLN antibody was synthesized at Fred Hutchinson Cancer Research Center (FHCRC; supplemental Methods) and conjugated to the fluorochrome phycoerythrin (Caprico Biotechnologies).
MSLN+ and MSLN− cell lines
Four MSLN− leukemia cell lines, K562 and Kasumi-1 (American Type Culture Collection) and Me-1 and MV4;11 (DSMZ), as well as 1 MSLN+ AML cell line, Nomo-1 (DSMZ), were purchased and used for MSLN expression and cytotoxicity assays.29 MSLN+ solid tumor lines H226 and PANC-1 (provided by Phil Greenberg, FHCRC) and OCVAR-3 (American Type Culture Collection) were used as positive controls in expression assays.29 Additional leukemia cell lines were created by lentiviral transduction of MSLN into K562 (K562-MSLN+), Kasumi-1 (Kasumi-1–MSLN+), Me-1 (Me-1–MSLN+), and MV4;11 (MV4;11-MSLN+), as well as CRISPR Nomo-1 MSLN knockout (Nomo-1–MSLNKO). They were sorted with a BD FACSAria II to obtain homogeneous populations (supplemental Methods).
In vitro cytotoxicity assays with ADCs
Three compounds were tested in vitro for MSLN-dependent cytotoxicity: (1) anetumab ravtansine (AR; Bayer Pharmaceuticals), an anti-MSLN antibody conjugated to payload DM4; (2) isotype control irrelevant monoclonal antibody conjugated to the same linker-payload as 1 (IC-AR; Bayer Pharmaceuticals); and (3) anti-MSLN–DGN462, an anti-MSLN monoclonal antibody conjugated to the DNA alkylating agent consisting of an indolino-benzodiazepine dimer (IBD; provided by ImmunoGen). In vitro cytotoxicity assays of AR and IC-AR were performed in MSLN+ and parental cell lines treated with ADCs (0.01 pM to 1 µM) in the presence of 20% human AB serum (Corning) and no azide/low endotoxin Fc receptor blocking agent (BD Pharmingen) for 30 minutes, washed twice with sterile phosphate-buffered saline, resuspended in fresh media per repository guidelines, incubated for 72 hours in duplicate, and then assessed using a Cell Titer-Glo Luminescent Cell Viability Assay (Promega). In vitro cytotoxicity assays of anti-MSLN–DGN462 experiments were performed as above and without an Fc-blocking agent.
In vivo treatment of MSLN+ leukemia xenografts with AR
NSG-B2m mice (stock number #010636; The Jackson Laboratory) were transplanted with 6 × 106 K562 or K562-MSLN+ cells via the tail vein, whereas NSG-SGM3 mice (stock number #03062; The Jackson Laboratory) were transplanted with 10 × 106 MV4;11-MSLN+ cells to produce cell line–derived xenografts, as described previously.30 At day 6 postinjection, mice were randomly assigned to 4 treatment groups: AR (5 mg/kg IV, every 3 days for 3 doses), IC-AR (5 mg/kg IV, every 3 days for 3 doses), chemotherapy (daunorubicin, 1.5 mg/kg IV daily for 3 days + cytarabine, 50 mg/kg intraperitoneally daily for 5 days), and no treatment (n = 6 per group for MSLN+ xenografts and n = 5 per group for MSLN− xenografts). Mice were monitored daily for humane end point criteria and euthanized per American Veterinary Medical Association guidelines.30 Experiments with patient-derived xenografts (PDXs) using an MSLN+ sample (NTPL-146) and an MSLN− sample (DF-2) were conducted as above utilizing NSG-SGM3 mice, with 3 × 106 cells injected per mouse. Treatment of the PDX with AR was initiated 23 days postinjection when the percentage of human leukemia cells in mouse PB was ≥0.1%, measured by MDF as previously described.30 The mice received 1 to 3 cycles of AR or IC-AR (supplemental Methods). Blood leukemia burden was assessed biweekly and then at increasing intervals up to 8 weeks, as well as at the time of euthanasia. Mouse studies were approved by the Nemours Institutional Animal Care and Use Committee.
Statistics
Correlation of clinical characteristics and outcome with MSLN expression was analyzed for 1038 patients treated on AAML1031 (supplemental Methods). Analyses of all in vitro and xenograft experiments was performed with Prism 7 (GraphPad; supplemental Methods).
Results
MSLN transcript expression in pediatric and adult AML
Comprehensive transcriptome profiling of pediatric AML (n = 200) initially identified MSLN as a highly expressed gene in a subset of patients. Subsequent transcriptome profiling of 1061 pediatric AML samples confirmed MSLN to be overexpressed in a subset of cases while absent from the majority of patients with AML. MSLN transcript expression varied significantly (range, 0-1222 TPM; median, 0.5) (Figure 1A). Defining MSLN overexpression (MSLN+) as ≥5 TPM, 36% of pediatric AML cases (n = 598) were MSLN+, with a median expression of 66 TPM (range, 5-1077.6). MSLN expression was virtually absent in normal hematopoiesis: median MSLN expression was 0.3 TPM in NBM (n = 68; range, 0.1-10.7) and 0.24 TPM in PB CD34+ cells (n = 16; range, 0.11-2.38) (Figure 1A). Evaluation of MSLN transcript expression in adult AML (n = 173) also revealed a wide range of values (0-703 TPM; median, 0.5 TPM). The prevalence of MSLN+ AML was 14% (n = 25; median, 127 TPM). MSLN expression in adult AML was significantly higher than in NBM (P < .0001). MSLN expression levels in pediatric and adult AML were significantly higher compared with normal hematopoietic samples (P < .001; supplemental Figure 2). Given the overexpression of MSLN in solid tumors, we compared MSLN expression in AML with MSLN+ solid tumors and found substantial overlap in the MSLN expression range (Figure 1B). We further evaluated expression of MSLN at relapse by analyzing matched diagnostic and relapse specimens from 139 patients with 20% blasts at both time points. We demonstrated a concordance rate of 90%; among MSLN+ patients at diagnosis, 76% retained MSLN expression at relapse, whereas only 4% of MSLN− patients at diagnosis acquired MSLN expression at relapse (Figure 1C).
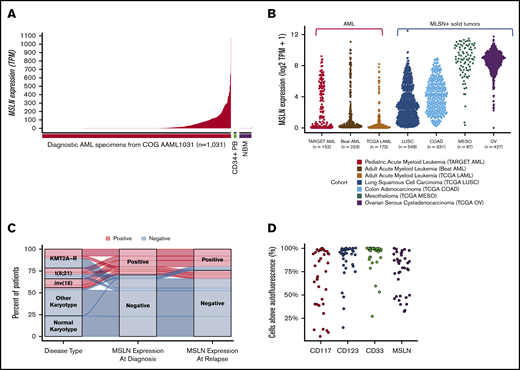
MSLN expression in pediatric and adult AML. (A) MSLN transcript expression was detected in a subset of pediatric AML cases (n = 1031) but was absent in NBM (n = 68) and CD34+ PB cells (n = 16), as determined by RNA-Seq. (B) MSLN expression in pediatric (TARGET cohort) and adult (TCGA and BEAT AML) AML patients compared with several MSLN+ solid tumors in patients from the TCGA cohort. (C) Concordance of MSLN transcript expression, positive vs negative, at diagnostic and relapse time points, according to karyotype (KMT2A-R, CBF, other, and normal karyotype). (D) Percentage of cells above autofluorescence, representing the percentage positivity on blasts, for some archetypal surface antigens in AML that are considered immunotherapeutic targets (CD117, CD33, CD123) and MSLN, showing similar distribution of heterogeneity of expression.
MSLN expression in pediatric and adult AML. (A) MSLN transcript expression was detected in a subset of pediatric AML cases (n = 1031) but was absent in NBM (n = 68) and CD34+ PB cells (n = 16), as determined by RNA-Seq. (B) MSLN expression in pediatric (TARGET cohort) and adult (TCGA and BEAT AML) AML patients compared with several MSLN+ solid tumors in patients from the TCGA cohort. (C) Concordance of MSLN transcript expression, positive vs negative, at diagnostic and relapse time points, according to karyotype (KMT2A-R, CBF, other, and normal karyotype). (D) Percentage of cells above autofluorescence, representing the percentage positivity on blasts, for some archetypal surface antigens in AML that are considered immunotherapeutic targets (CD117, CD33, CD123) and MSLN, showing similar distribution of heterogeneity of expression.
Verification of MSLN transcript expression by qRT-PCR demonstrated that it ranged from 0 to 3993 copies per 1000 copies of GUSB in pediatric AML (n = 619), whereas it ranged from 0 to 1388 copies per 1000 copies of GUSB in adult AML (n = 41) and from 0.5 to 3.7 copies per 1000 copies of GUSB in NBM (n = 16) (supplemental Figure 2). MSLN expression values by qRT-PCR and RNA-Seq demonstrated a strong correlation (Spearman nonparametric r = +0.86; P < .0001). When defining MSLN positivity as ≥50 copies of MSLN per 1000 copies of GUSB, 29% (n = 180) of pediatric AML cases and 29% (n = 12) of adult AML cases were MSLN+.
Cell surface MSLN expression in AML
Prospective screening of diagnostic specimens from 138 consecutive AML patients for MSLN expression by MDF detected surface expression on the blasts in 29% (n = 40) of patients. Heterogeneity of MSLN expression was similar to that observed for other archetypal AML cell surface antigens (eg, CD117, CD33, CD123; Figure 1D). Evaluation of NBM samples demonstrated the absence of any detectable MSLN. In all MSLN+ cases, MSLN expression was confined to the leukemic blasts and was absent from normal hematopoietic cells (Figure 2). Median MFI was 34.7 (range, 9.28-498) in the MSLN+ cohort vs 5.84 (range, 2.32-14.15) in the MSLN− cohort (P < .0001; supplemental Figure 2). Among the MSLN+ cohort, expression was heterogeneous across a subset of cases (38%; n = 15), with 30% to 70% of the blast population expressing MSLN.
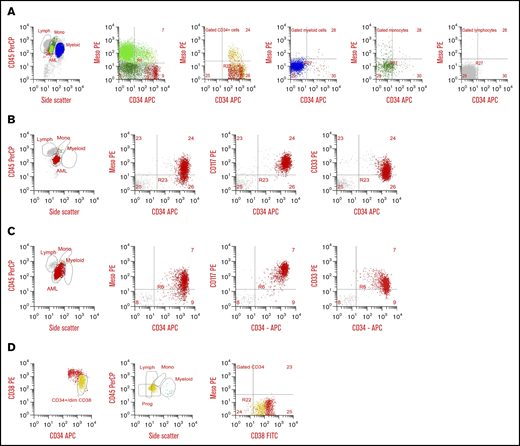
Cell surface MSLN expression in AML. (A) Flow plots from a 5-year-old with MSLN+ AML with predominantly CD34−/MSLN+ leukemia. First plot with CD45/side scatter (SSC) distribution showing leukemic blasts (bright green), normal monocytes (dark green), myeloid cells (blue), and normal lymphocytes (gray). In the second plot, AML is shown in bright green, monocytes are dark green, and CD34+ cells are red. The AML is predominantly CD34− but both CD34− and CD34+ subset express MSLN. The third plot is gated on CD34+ cells, with CD34+/MSLN+ leukemic blasts in yellow and CD34+ normal progenitor cells in red. The fourth, fifth, and sixth plots show normal myeloid cells (blue), normal monocytes (green), and normal lymphocytes (gray), respectively, none of which express MSLN. (B) Flow plots from a 12-year-old with MSLN+/CD34+ AML. First plot with CD45/SSC distribution showing leukemic blasts (red), a few normal monocytes (green), and normal lymphocytes (gray). In the second plot the CD34+ blasts demonstrate MSLN expression. Third plot confirms the myeloid nature of CD34+/heterogenous CD117+ abnormal blasts (red), and normal lymphocytes (gray). The fourth plot confirms myeloid nature of the CD34+/CD33+ blasts (red), with lymphocytes (gray). (C) Flow plots from a 17-year-old patient with MSLN+/CD34+ AML. First plot with CD45/SSC distribution showing leukemic blasts (red), and few normal myeloid and normal lymphocytes (orange and gray, respectivly). Second plot shows heterogeneous MSLN expression on abnormal CD34+ myeloblasts (red) and normal lymphocytes (gray). The third plot confirms the myeloid nature of CD34+/CD117+ abnormal blasts (red) and normal lymphocytes (gray). (D) Normal CD34+ progenitor cells from an 18-year-old are negative for MSLN expression. The first plot shows CD34+ cells with early progenitors with bright CD34+/dim CD38+ expression in yellow, the second plot shows the characteristic position of these early progenitors (yellow) by CD45 and SSC, and the third plot shows there is no MSLN expression on any normal CD34+ cells, either early progenitors (yellow) or uncommitted progenitors (red). APC, allophycocyanin; FITC, fluorescein isothiocyanate; Lymph, lymphocytes; Meso, mesothelin; Mono, monocytes; PE, phycoerythrin; PerCP, peridinin-chlorophyll-protein complex; Prog, progenitors.
Cell surface MSLN expression in AML. (A) Flow plots from a 5-year-old with MSLN+ AML with predominantly CD34−/MSLN+ leukemia. First plot with CD45/side scatter (SSC) distribution showing leukemic blasts (bright green), normal monocytes (dark green), myeloid cells (blue), and normal lymphocytes (gray). In the second plot, AML is shown in bright green, monocytes are dark green, and CD34+ cells are red. The AML is predominantly CD34− but both CD34− and CD34+ subset express MSLN. The third plot is gated on CD34+ cells, with CD34+/MSLN+ leukemic blasts in yellow and CD34+ normal progenitor cells in red. The fourth, fifth, and sixth plots show normal myeloid cells (blue), normal monocytes (green), and normal lymphocytes (gray), respectively, none of which express MSLN. (B) Flow plots from a 12-year-old with MSLN+/CD34+ AML. First plot with CD45/SSC distribution showing leukemic blasts (red), a few normal monocytes (green), and normal lymphocytes (gray). In the second plot the CD34+ blasts demonstrate MSLN expression. Third plot confirms the myeloid nature of CD34+/heterogenous CD117+ abnormal blasts (red), and normal lymphocytes (gray). The fourth plot confirms myeloid nature of the CD34+/CD33+ blasts (red), with lymphocytes (gray). (C) Flow plots from a 17-year-old patient with MSLN+/CD34+ AML. First plot with CD45/SSC distribution showing leukemic blasts (red), and few normal myeloid and normal lymphocytes (orange and gray, respectivly). Second plot shows heterogeneous MSLN expression on abnormal CD34+ myeloblasts (red) and normal lymphocytes (gray). The third plot confirms the myeloid nature of CD34+/CD117+ abnormal blasts (red) and normal lymphocytes (gray). (D) Normal CD34+ progenitor cells from an 18-year-old are negative for MSLN expression. The first plot shows CD34+ cells with early progenitors with bright CD34+/dim CD38+ expression in yellow, the second plot shows the characteristic position of these early progenitors (yellow) by CD45 and SSC, and the third plot shows there is no MSLN expression on any normal CD34+ cells, either early progenitors (yellow) or uncommitted progenitors (red). APC, allophycocyanin; FITC, fluorescein isothiocyanate; Lymph, lymphocytes; Meso, mesothelin; Mono, monocytes; PE, phycoerythrin; PerCP, peridinin-chlorophyll-protein complex; Prog, progenitors.
Evaluation of soluble MSLN in AML
We evaluated the diagnostic serum of 337 pediatric patients and 43 adult patients to determine the presence and levels of serum-soluble MSLN (ss-MSLN) in AML. Using the Mesomark positivity cutoff ≥ 1.5 nM, which has been established for MSLN+ solid tumors, 25% of pediatric AML cases (n = 86) and 33% of adult AML cases (n = 14) had high levels of ss-MSLN (Figure 3A). Comparison of ss-MSLN ELISA data with transcriptome data in 122 pediatric AML cases found a direct correlation between transcript and ss-MSLN expression (Spearman r = +0.57; P < .0001; Figure 3B). Comparison of ELISA and transcriptome data for detecting MSLN+ showed a Cohen’s κ of 0.76 for test agreement, and the area under the curve was 0.83 (supplemental Figure 4). ss-MSLN ELISA demonstrated a specificity of 97% (95% confidence interval [CI], 91.0-99.7) and a sensitivity of 75% (95% CI, 59.7-86.8) at the ≥1.5-nM cutoff, establishing that elevated ss-MSLN is highly indicative of MSLN transcript overexpression in diagnostic AML samples (supplemental Table 1). A similar agreement between ss-MSLN ELISA and qRT-PCR assays was observed (n = 143; κ = 0.73; area under the curve, 0.82). These findings suggest that test performance in AML is comparable to Mesomark in mesothelioma.

Elevated ss-MSLN in patients with MSLN+AML. (A) ss-MSLN levels at diagnosis in 337 pediatric patients and 43 adult AML patients, measured by ELISA, comparable to Mesomark. Using a positivity threshold ≥1.5 nM, 25% of pediatric patients and 33% of adult AML patients were positive for soluble MSLN. (B) ss-MSLN levels correlate with MSLN transcript levels detected by RNA-Seq (Spearman r = +0.57; P < .0001). Thresholds of MSLN+ ≥1.5 nM and ≥5 TPM are illustrated. (C) ss-MSLN levels in 39 AML patients who were positive for soluble MSLN at diagnosis (Dx) and had a paired serum sample collected at the EOI chemotherapy in the setting of an MRD-negative remission.
Elevated ss-MSLN in patients with MSLN+AML. (A) ss-MSLN levels at diagnosis in 337 pediatric patients and 43 adult AML patients, measured by ELISA, comparable to Mesomark. Using a positivity threshold ≥1.5 nM, 25% of pediatric patients and 33% of adult AML patients were positive for soluble MSLN. (B) ss-MSLN levels correlate with MSLN transcript levels detected by RNA-Seq (Spearman r = +0.57; P < .0001). Thresholds of MSLN+ ≥1.5 nM and ≥5 TPM are illustrated. (C) ss-MSLN levels in 39 AML patients who were positive for soluble MSLN at diagnosis (Dx) and had a paired serum sample collected at the EOI chemotherapy in the setting of an MRD-negative remission.
Because Mesomark is used clinically for monitoring mesothelioma patients during therapy, we tested the ability of ss-MSLN to indicate disease remission at the end of induction (EOI) chemotherapy. In a cohort of 39 pediatric MSLN+ AML patients with paired serum samples at diagnosis and EOI chemotherapy, 38 (97%) were negative for soluble MSLN at EOI chemotherapy (Figure 3C), with 37 of those patients being measurable residual disease (MRD) negative. Among the subset with paired ss-MSLN and outcome data (n = 29), the 3 patients with soluble MSLN > 1 nM at EOI chemotherapy ultimately relapsed, despite their MRD-negative remission status at EOI chemotherapy.
Biologic and clinical correlates of MSLN expression in AML
Using the cutoff of ≥5 TPM for MSLN+ AML and <5 TPM for MSLN− AML, we analyzed 1038 pediatric patients enrolled on AAML1031 to evaluate the association between MSLN expression and clinical characteristics and outcome. MSLN expression was not associated with sex, ethnicity, or the number of white blood cells or peripheral blasts at diagnosis. However, MSLN expression was associated with age and was strongly associated with cytogenetic and molecular subgroups (Table 1). MSLN+ disease was detected in 62% of patients with KMT2A rearrangements (KMT2A-R; P < .001) and in 72% of those with core binding factor (CBF) AML [88% with inv(16)/t(16;16) and 60% with t(8;21); P < .001 for each group; Table 1]. MSLN+ expression was significantly associated with extramedullary disease (EMD), with EMD occurring in 27.8% of MSLN+ patients vs 18.8% of MSLN− patients (P = .001; Table 1). Evaluation of CBF and KMT2A-R patients did not demonstrate any significant differences in EMD according to MSLN expression (supplemental Table 2). MSLN expression was rare or absent among FLT3-ITD, NPM1, and CEBPα-mutated AML (Table 1). In a multivariable analysis, there was no association between MSLN and overall survival or event-free survival (P = .384 and .412, respectively). Analysis of KMT2A-R and CBF subgroups also did not demonstrate any association between MSLN expression and outcome (supplemental Table 3). Given the association between MSLN+ AML and EMD, we evaluated relapsed/refractory cases (n = 486) and found that, among the MSLN+ cohort (n = 169), 40.9% (n = 69) had ≥1 site of EMD compared with 18.7% of MSLN− patients (60/317; P < .001).
Clinical and biologic characteristics of MSLN−and MSLN+children and young adult patients treated on AAML1031
| Characteristic | MSLN− (<5 TPM), n = 679 | MSLN+ (≥5 TPM), n = 359 | P |
|---|---|---|---|
| Males | 344 (50.7) | 193 (53.8) | .342 |
| Age, median (range), y | 11.1 (0.04-29.5) | 8.3 (0-28.3) | .002 |
| CNS disease classification | |||
| CNS1 | 488 (73.3) | 206 (60.4) | <.001 |
| CNS2 | 126 (19.1) | 90 (26.4) | .008 |
| CNS3 | 50 (7.6) | 45 (13.2) | .004 |
| Non-CNS EMD present | 84 (12.4) | 61 (17) | .042 |
| Any EMD (non-CNS + CNS3) | 125 (18.8) | 95 (27.8) | .001 |
| Diagnostic WBC count, median (range), ×103/μL | 19.9 (0.6-918.5) | 28.1 (0.6-712.7) | .116 |
| Bone marrow blasts, median (range), % | 68 (0-100) | 70 (0-100) | .074 |
| Peripheral blasts, median (range), % | 36 (0-100) | 40 (0-99) | .791 |
| CEBPα positive | 62 (9.1) | 0 (0) | <.001 |
| NPM1 positive | 91 (13.4) | 5 (1.4) | <.001 |
| FLT3-ITD positive | 156 (23) | 12 (3.3) | <.001 |
| Cytogenetics | |||
| Normal | 257 (36.9) | 10 (2.8) | <.001 |
| inv(16)/t(16;16) | 12 (1.8) | 89 (24.9) | <.001 |
| t(8;21) | 57 (8.5) | 86 (24) | <.001 |
| 11q23/KMT2A rearrangements | 87 (13) | 144 (40.2) | <.001 |
| Monosomy 5/del5q | 10 (1.5) | 0 (0) | .018 |
| Monosomy 7 | 18 (2.7) | 1 (0.3) | .006 |
| Trisomy 8 | 50 (7.5) | 13 (3.6) | .015 |
| Other abnormalities | 159 (23.7) | 14 (3.9) | <.001 |
| Complete remission at end of induction I | 479 (72.1) | 269 (82.3) | <.001 |
| MRD < 0.1% at end of induction I | 406 (63.7) | 301 (87.8) | <.001 |
| Characteristic | MSLN− (<5 TPM), n = 679 | MSLN+ (≥5 TPM), n = 359 | P |
|---|---|---|---|
| Males | 344 (50.7) | 193 (53.8) | .342 |
| Age, median (range), y | 11.1 (0.04-29.5) | 8.3 (0-28.3) | .002 |
| CNS disease classification | |||
| CNS1 | 488 (73.3) | 206 (60.4) | <.001 |
| CNS2 | 126 (19.1) | 90 (26.4) | .008 |
| CNS3 | 50 (7.6) | 45 (13.2) | .004 |
| Non-CNS EMD present | 84 (12.4) | 61 (17) | .042 |
| Any EMD (non-CNS + CNS3) | 125 (18.8) | 95 (27.8) | .001 |
| Diagnostic WBC count, median (range), ×103/μL | 19.9 (0.6-918.5) | 28.1 (0.6-712.7) | .116 |
| Bone marrow blasts, median (range), % | 68 (0-100) | 70 (0-100) | .074 |
| Peripheral blasts, median (range), % | 36 (0-100) | 40 (0-99) | .791 |
| CEBPα positive | 62 (9.1) | 0 (0) | <.001 |
| NPM1 positive | 91 (13.4) | 5 (1.4) | <.001 |
| FLT3-ITD positive | 156 (23) | 12 (3.3) | <.001 |
| Cytogenetics | |||
| Normal | 257 (36.9) | 10 (2.8) | <.001 |
| inv(16)/t(16;16) | 12 (1.8) | 89 (24.9) | <.001 |
| t(8;21) | 57 (8.5) | 86 (24) | <.001 |
| 11q23/KMT2A rearrangements | 87 (13) | 144 (40.2) | <.001 |
| Monosomy 5/del5q | 10 (1.5) | 0 (0) | .018 |
| Monosomy 7 | 18 (2.7) | 1 (0.3) | .006 |
| Trisomy 8 | 50 (7.5) | 13 (3.6) | .015 |
| Other abnormalities | 159 (23.7) | 14 (3.9) | <.001 |
| Complete remission at end of induction I | 479 (72.1) | 269 (82.3) | <.001 |
| MRD < 0.1% at end of induction I | 406 (63.7) | 301 (87.8) | <.001 |
Unless otherwise noted, data are n (%).
CNS1, no blasts identified; CNS2, blasts present on cytospin with white blood cells (WBC) <5 or blasts present on cytospin with WBC ≥5 and traumatic tap; CNS3, blasts present on cytospin with WBC ≥5 and atraumatic tap.
In an effort to explore the underlying mechanism of MSLN expression in AML, we interrogated the available genome, epigenome, and transcriptome data (TARGET and TCGA) comparing those with and without MSLN transcript expression. Available miRNA data from patients with and without MSLN did not identify any miRNAs whose expression correlated with MSLN transcript expression. Evaluation of the DNA methylation data from those with MSLN expression demonstrated an inverse association between MSLN promoter methylation and MSLN transcript expression levels (Pearson’s r = −0.645; P < .001; supplemental Figure 5), demonstrating that MSLN expression in AML may be the result of epigenomic alterations in AML. Analysis of a cohort of TCGA patients (n = 155) did not find any association between mutations of genes involved in DNA methylation (DNMT3A, IDH1/2, TET1/2) and MSLN expression (P > .2 for all).
Cytotoxicity of MSLN-targeted ADCs
To assess the preclinical efficacy of MSLN-directed therapies in AML, we conducted studies of the anti-MSLN ADC AR, as well as a newly devised ADC (anti-MSLN–DGN462). We tested the ADCs in cells naturally expressing MSLN+ (Nomo-1 and its CRISPR-deleted MSLN- counterpart Nomo-1 MSLNKO), as well as in cell lines that were engineered to express MSLN (K562-MSLN+, Kasumi-1–MSLN+, Me-1–MSLN+, MV4;11-MSLN+) and their MSLN parental cell lines (K562, Kasumi-1, Me-1, and MV4;11; supplemental Table 4). Target-dependent cytotoxicity of AR was observed in MV4;11-MSLN+ and K562-MSLN+ cells (half-maximal inhibitory concentration [IC50] = 1.5 nM and 3.7 nM, respectively), with little to no cytotoxicity observed in control conditions, including IC-AR treatment and treatment of the MSLN− parental lines (P < .0001, between IC50 following treatment with AR compared with both MV4;11-MSLN+ and K562-MSLN+ cell lines; Figure 4A; supplemental Figure 6). Three MSLN+ leukemia cell lines (Nomo-1, Kasumi-1–MSLN+, Me-1–MSLN+) were not sensitive to AR (supplemental Figure 6). To examine whether MSLN targeting in leukemia could be improved using an ADC with a DNA-damaging payload, we evaluated the efficacy of anti-MSLN–DGN462, which demonstrated target-dependent cytotoxicity in MV4;11-MSLN+, K562-MSLN+, Nomo-1, Kasumi-1, and Me-1-MSLN+ cells, with IC50 = 50 pM, 1.2 nM, 0.27 nM, 1 nM, and 7.3 nM, respectively (Figure 4B-C; supplemental Figure 6).
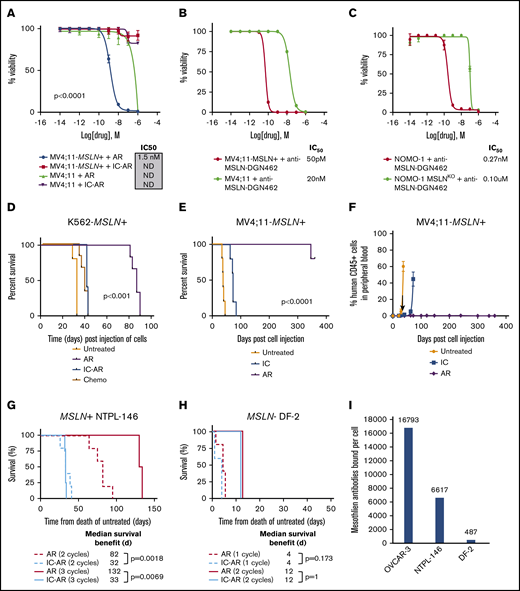
In vitro and in vivo cytotoxicity of MSLN-targeted ADCs in MSLN+leukemia cell lines. (A) In vitro cytotoxicity of AR in MV4;11-MSLN+ cell lines and IC50 values. Controls are IC-AR and treatment of the parental (MSLN−) lines. In vitro cytotoxicity of the ADC anti-MSLN–DGN462 with an indolino-benzodiazepine dimer payload in MV4;11-MSLN+ and MV4;11 parental cells (B) and Nomo-1 parental cells and Nomo-1–MSLNKO cells (C). (D) Kaplan-Meier survival plots of K562-MSLN+ cell–xenografted mice treated with AR compared with IC-AR, chemotherapy (Chemo), and no treatment. (E) Kaplan-Meier survival plots of MV4;11-MSLN+ xenografted mice treated with AR, along with controls: IC-AR (IC) and untreated. (F) PB leukemia burden was assessed in MV4;11-MSLN+ mice by flow cytometry. (G) Treatment of MSLN+ PDX NTPL-146 with AR resulted in a dose-dependent improvement in median survival with respect to untreated mice. Mice treated with AR vs IC-AR for 2 cycles (dashed lines; n = 5 per group) experienced a median survival of 82 days vs 32 days (P = .0018) and mice treated for 3 cycles (solid lines; n = 4 per group) had a median survival of 132 days vs 33 days, respectively (P = .0069; n = 4 per group). (H) Treatment of the MSLN− PDX DF-2 with AR did not demonstrate any target-dependent efficacy compared with untreated IC-AR mice. Mice treated with AR vs IC-AR for 1 cycle (dotted lines; n = 4 per group) experienced an identical median survival of 4 days (P = 1.0) and mice treated for 2 cycles (solid lines; n = 5 per group) had identical median survival of 12 days, respectively (P = .173; n = 4 per group). (I) Quantification of cell surface mesothelin expression using BD Quantibrite, as measured by antibodies bound per cell in the MSLN+ ovarian cancer cell line OCVAR-3 used as positive control and the PDX models NTPL-146 and DF2. ND, IC50 could not be determined with 95% CIs.
In vitro and in vivo cytotoxicity of MSLN-targeted ADCs in MSLN+leukemia cell lines. (A) In vitro cytotoxicity of AR in MV4;11-MSLN+ cell lines and IC50 values. Controls are IC-AR and treatment of the parental (MSLN−) lines. In vitro cytotoxicity of the ADC anti-MSLN–DGN462 with an indolino-benzodiazepine dimer payload in MV4;11-MSLN+ and MV4;11 parental cells (B) and Nomo-1 parental cells and Nomo-1–MSLNKO cells (C). (D) Kaplan-Meier survival plots of K562-MSLN+ cell–xenografted mice treated with AR compared with IC-AR, chemotherapy (Chemo), and no treatment. (E) Kaplan-Meier survival plots of MV4;11-MSLN+ xenografted mice treated with AR, along with controls: IC-AR (IC) and untreated. (F) PB leukemia burden was assessed in MV4;11-MSLN+ mice by flow cytometry. (G) Treatment of MSLN+ PDX NTPL-146 with AR resulted in a dose-dependent improvement in median survival with respect to untreated mice. Mice treated with AR vs IC-AR for 2 cycles (dashed lines; n = 5 per group) experienced a median survival of 82 days vs 32 days (P = .0018) and mice treated for 3 cycles (solid lines; n = 4 per group) had a median survival of 132 days vs 33 days, respectively (P = .0069; n = 4 per group). (H) Treatment of the MSLN− PDX DF-2 with AR did not demonstrate any target-dependent efficacy compared with untreated IC-AR mice. Mice treated with AR vs IC-AR for 1 cycle (dotted lines; n = 4 per group) experienced an identical median survival of 4 days (P = 1.0) and mice treated for 2 cycles (solid lines; n = 5 per group) had identical median survival of 12 days, respectively (P = .173; n = 4 per group). (I) Quantification of cell surface mesothelin expression using BD Quantibrite, as measured by antibodies bound per cell in the MSLN+ ovarian cancer cell line OCVAR-3 used as positive control and the PDX models NTPL-146 and DF2. ND, IC50 could not be determined with 95% CIs.
We further assessed the in vivo efficacy of AR in MSLN+ leukemia in cell line–derived xenograft and PDX models of MSLN+ AML. K562-MSLN+ xenografts treated with AR had significantly prolonged survival (median, 87 days) compared with IC-AR treatment, chemotherapy treatment (daunorubicin and cytarabine), and no treatment, with median survival times of 41, 38, and 32 days, respectively (P < .0001; Figure 4D). In contrast, parental K562 xenografts treated with AR had similar survival compared with IC-AR treatment or no treatment, with median survival of 39, 41, and 32 days, respectively (supplemental Figure 6). MV4;11-MSLN+ xenografts treated with AR uniformly had prolonged survival > 340 days, whereas those treated with IC-AR or left untreated had a median survival of 72 and 38 days, respectively, with symptomatic leukemia co-occurring with increasing blood leukemia burden (P < .0001; Figure 4E). Additionally, MV4;11-MSLN+ xenografts treated with AR had <1% peripheral leukemia burden throughout the prolonged posttreatment monitoring period (Figure 4F). Treatment of the MSLN+ PDX NTPL-146 with AR resulted in a median survival of 82 days compared with 32 days (P = .0018) for mice treated with 2 cycles of IC-AR and 132 vs 33 days, respectively, for mice treated with 3 cycles of IC-AR (P = .0069; Figure 4G; supplemental Figure 6). In contrast, treatment of the MSLN− PDX DF-2 with AR and IC-AR resulted in identical median survivals of 12 days for 1 treatment cycle (P = 1.0) and 4 days for 2 cycles (P = .173; Figure 4H).
Discussion
Utilizing large-scale next-generation sequencing efforts, we identified MSLN, a known targetable cell surface protein in solid tumors, to be a highly overexpressed AML-restricted transcript in a significant proportion of AML, regardless of age. We verified expression by qRT-PCR, MDF, and a plasma-soluble MSLN assay. Given the high expression in AML with low/absent expression in normal hematopoiesis and paucity of expression in most other tissues, MSLN is an ideal therapeutic target in AML because leukemic cells may be targeted with virtually no hematopoietic toxicity. We demonstrated proof-of-principle target-dependent killing of MSLN+ leukemia in vitro and in vivo, with a significant survival benefit in MSLN+ AML xenografts using AR, which is under clinical investigation in solid tumors. We further demonstrated that MSLN targeting with ADCs might be improved in AML by utilizing a DNA-damaging payload.
MSLN is a potentially valuable marker of disease in AML, as in solid tumors. Detection of cell surface expression by MDF and qRT-PCR to detect transcript overexpression have clinical potential as diagnostic and MRD assays in MSLN+ AML. Molecular MRD assays in AML have been shown to be powerful tools for disease monitoring and can complement MDF-based methods to provide superior disease detection and prognostic capability.31-33 Blood testing for ss-MSLN, similar to Mesomark in mesothelioma, presents a potential novel and less invasive means of therapeutic monitoring in AML. In our study, we demonstrated good intertest agreement of serum and transcript MSLN measurement for detecting MSLN+ AML at diagnosis. We observed that ss-MSLN in MSLN+ AML generally fell into the normal range at EOI chemotherapy, in accordance with achieving remission, suggesting that postinduction detection of soluble MSLN may correlate with subsequent relapse. Our findings support testing MSLN expression in prospective clinical trials to further define optimal methods for detection and quantification in AML.
Overexpression of MSLN on AML blasts is unexpected, given its virtual absence in normal hematopoietic cells and the seeming lack of a relationship between AML and normal MSLN-expressing mesothelial cells. Our findings build on a prior smaller series that identified overexpression of MSLN in a subset of pediatric AML cases.34 We found MSLN expressed on AML blasts in patients across the age spectrum. The breadth and depth of sequencing modalities and the large cohort size allowed comprehensive evaluation of the association between MSLN overexpression in pediatric AML and possible etiologies. We show that MSLN overexpression was strongly associated with KMT2A-R and CBF AML. Although MSLN overexpression was associated with favorable complete remission rate, this was likely due to the overrepresentation of CBF patients who experience favorable responses to therapy,35 because multivariate analyses among the CBF and KMT2A-R subgroups demonstrated that MSLN expression was not associated with outcome. We observed a striking association between MSLN expression and EMD, and we hypothesize that this is due to more than just the overlap with the KMT2A-R and CBF subtypes, which have a higher prevalence of EMD compared with other AML subtypes.36-38 Mesothelial cells are implicated in processes of cell-cell adhesion, loss of adhesion, and migratory properties.39-41 Further, MSLN has been reported to be involved in cell adhesion in ovarian carcinoma.42 Our findings suggest that further work is needed to explore this association and potential functional implications of MSLN in AML, because expression of MSLN may be implicated in EMD development. We also found a strong association between MSLN overexpression and promoter hypomethylation. Epigenetic dysregulation is a common paradigm in AML pathophysiology, including effects on t(8;21) and KMT2A-R AML.43,44 Treatment with epigenetic-modifying agents has been proposed to modify MSLN expression of MSLN+ solid tumors45-47 ; additional work is needed to understand the regulation of MSLN expression in AML and whether it also may be subjected to therapeutic manipulation.
In this study, we demonstrate that AR has activity in vitro and in vivo against MSLN+ AML. AR conferred target-dependent in vitro cytotoxicity, as well as resulted in significant survival benefits in MSLN+ AML xenografts, because treatment with AR resulted in a dose-dependent improvement in median survival compared with no treatment. Although not all MSLN+ AML cell lines were sensitive to AR, we hypothesize that this may be due, in part, to its payload, DM4, a tubulin inhibitor that is dependent on cell cycling for effect. Therefore, we evaluated anti-MSLN–DGN462, which utilizes an alkylator payload (IBD), a mechanism of action that has shown to be effective in AML and is suggested to be uniquely effective in quiescent malignant cells.48-52 This ADC exhibited target-dependent cytotoxicity against a broader range of MSLN+ cell lines compared with AR, suggesting that more optimized payloads for AML may increase therapeutic efficacy. Although a prior study in MSLN+ AML with an MSLN-targeted immunotoxin failed to demonstrate efficacy,53 our studies of MSLN-targeted ADCs support further clinical investigation of these agents in MSLN+ AML.
We performed the first preclinical evaluation of AR in AML and demonstrate that MSLN-targeted agents are a promising therapeutic strategy. Efforts in solid tumors might be leveraged to advance MSLN targeting in AML, because AR has been studied in early-phase clinical trials in adults with advanced MSLN+ tumors.54,55 Phase I evaluation of AR reported low rates of serious treatment-emergent adverse events, with rare and reversible significant hematologic events.55 Many patients experienced stable disease in response to AR; however, objective responses were rare.55 Although targeting of solid tumors with ADC monotherapy has proven challenging in many instances (mediated, in part, by expression level, access to tumor, and microenvironment), a number of hematologic malignancies have been successfully targeted with ADCs.55-60 Therefore, results using AR in solid tumors should not be directly applied to AML. MSLN-directed therapies can avoid the on-target/off-tumor toxicity observed with other immunotherapeutic targets used in AML (ie, CD33). This shared antigen expression among AML and normal hematopoietic cells is a dose-limiting side effect of ADCs in AML.1,48,49,61 One potential drawback of MSLN-targeted ADCs is that ss-MSLN could act as a sink for the drug, preventing it from binding to MSLN on leukemic cells. To diminish the source of soluble MSLN, clinical trials of MSLN-targeted ADCs in AML could consider a cytoreduction phase prior to ADC dosing. Antigen load at diagnosis due to a high white blood cell count is a concern for ADCs; thus, they may be most effective when administered as combination therapy in hematologic malignancies.62,63 Immunotherapeutic targeting of MSLN in AML could also include chimeric antigen receptor T cells and T-cell receptor therapies as little off-tumor/on-target toxicity has been detected in clinical trials utilizing these strageies in MSLN+ solid tumors.14
MSLN expression is observed across the age spectrum in AML but is absent from normal hematopoietic precursors; the progress made in MSLN+ solid tumors with an array of targeted and immunotherapeutic strategies positions MSLN to be an impactful new therapeutic target in AML. Our studies support further work to prospectively evaluate the role of MSLN overexpression, optimal detection methods, and the development of clinical trials evaluating MSLN-directed therapeutic strategies in AML.
Transcriptome data for patients used in this study have been deposited in the Database of Genotypes and Phenotypes (https://www.ncbi.nlm.nih.gov/gap/; accession number phs000465.v19.p8; TARGET: Acute Myeloid Leukemia). This is a substudy of the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project (accession number phs000218.v22.p8). Data are also accessible at the National Cancer Institute’s Genomic Data Commons Portal (https://portal.gdc.cancer.gov/) under TARGET-AML.
Data sharing requests should be sent to Katherine Tarlock (katherine.tarlock@seattlechildrens.org).
Acknowledgments
The authors thank Bayer HealthCare Pharmaceuticals, Inc. for providing AR and the isotype control for AR at no cost for this project. They also acknowledge the MDACC for the contribution of samples for this analysis.
This work was supported by the St. Baldrick’s Foundation, a St. Baldrick’s Scholar Award (K.T.), a St. Baldrick’s Consortium Grant (S.M.), TARGET Pediatric AML (S.M.), the Leukemia and Lymphoma Society (6558-18 [S.M. and E.A.K] and 6604-20 [S.M.]), National Institutes of Health, National Cancer Institute Research Training and Career Development grants K12-CA076930 (A.J.K.) and T32-CA009351 (A.J.K.), and Research Grant Program R01-CA114563-10 (S.M.), Department of Health and Human Services HHSN-261200800001E (S.M.), the Andrew McDonough B+ Foundation (S.M.), the American Society of Clinical Oncology Conquer Cancer Foundation (A.J.K.), a COG Chair’s grant (U10-CA098543) (S.M.), the Children’s Oncology Group Foundation (K.T.), Hyundai Hope on Wheels (S.M.), the National Cancer Institute’s National Clinical Trials Network Statistics and Data Center (U10-CA180899) (S.M. and T.A.A.), Project Stella (S.M.), the Rally Foundation/Truth 365 (A.J.K.), and Leukemia Research Foundation of Delaware (S.P.B. and A.G.). This study used the computational infrastructure of FHCRC Scientific Computing, which is funded by the Office of Research Infrastructure Programs grant S10OD028685.
Authorship
Contribution: A.J.K., S.P.B., T. Tang, Q.H.L., L.P., A.G., and C.K.C. performed experiments; A.J.K., S.P.B., R.E.R., T. Tang, Q.H.L., T.A.A., R.B.G., L.E.B., L.P., M.X., A.R.L, J.L.S., T. Triche, and K.T. analyzed data; A.J.K., S.P.B., A.R.L., J.L.S., and K.T. prepared figures; A.J.K., K.T., E.A.K., and S.M. designed the experiments; M.R.L., C.C., and S.M.K. provided general scientific guidance; A.J.K., K.T., and S.M. wrote the manuscript; and all authors reviewed the manuscript prior to submission.
Conflict-of-interest disclosure: A.J.K. is an employee of and has equity ownership in Bristol Myers Squibb. M.R.L. is an employee of and has equity ownership in Hematologics Inc. L.E.B. and L.P. are employees of Hematologics Inc. C.C. has received research funding from, as well as travel funding to participate in an advisory board meeting for, Nektar Therapeutics. Some of C.C.’s current research is also funded by Nektar Therapeutics (through the current principal investigator who is not an author of this manuscript); the advisory board and current research are not related to this manuscript. The remaining authors declare no competing financial interests.
Correspondence: Katherine Tarlock, Seattle Children’s Hospital, Department of Hematology/Oncology, 4800 Sand Point Way NE, MB.8.501, Seattle, WA 98105; e-mail: katherine.tarlock@seattlechildrens.org.

