

Key Points
New target genes for γ-globin induction identified among all current potential therapeutic agents in an erythroid-biased CRISPR screen.
Top novel drug candidates and pathways for fetal hemoglobin induction examined and validated.

Abstract
Human γ-globin is predominantly expressed in fetal liver erythroid cells during gestation from 2 nearly identical genes, HBG1 and HBG2, that are both perinatally silenced. Reactivation of these fetal genes in adult red blood cells can ameliorate many symptoms associated with the inherited β-globinopathies, sickle cell disease, and Cooley anemia. Although promising genetic strategies to reactivate the γ-globin genes to treat these diseases have been explored, there are significant barriers to their effective implementation worldwide; alternatively, pharmacological induction of γ-globin synthesis could readily reach the majority of affected individuals. In this study, we generated a CRISPR knockout library that targeted all erythroid genes for which prospective or actual therapeutic compounds already exist. By probing this library for genes that repress fetal hemoglobin (HbF), we identified several novel, potentially druggable, γ-globin repressors, including VHL and PTEN. We demonstrate that deletion of VHL induces HbF through activation of the HIF1α pathway and that deletion of PTEN induces HbF through AKT pathway stimulation. Finally, we show that small-molecule inhibitors of PTEN and EZH induce HbF in both healthy and β-thalassemic human primary erythroid cells.
Introduction
The β-globinopathies (sickle cell disease [SCD] and Cooley anemia [β-thalassemia major]) comprise the most common inherited monogenic diseases.1-3 Increased levels of fetal hemoglobin (HbF) in adult red blood cells inversely correlate with SCD disease severity.4,5 To date, hydroxyurea is the most prevalent US Food and Drug Administration-approved drug used for the treatment of SCD that will induce HbF,6 but response to hydroxyurea varies among individuals. For this reason, development of alternative therapeutic approaches to HbF induction will benefit this globally underresourced population.
In this study, we designed a custom CRISPR knockout library to identify genes (1) that function to repress γ-globin expression in adult red blood cells and (2) for which modifying compounds have been characterized. By probing this library in human umbilical cord blood-derived erythroid progenitor 2 (HuDEP2) cells, we identified several novel genes encoding proteins that repress γ-globin expression. Among these genes, deletion of VHL induces HbF through the activation of HIF1α, whereas deletion of PTEN induces HbF by activation of the AKT signaling pathway. Furthermore, pharmacological inhibitors of both PTEN and EZH are shown to significantly induce HbF.
Methods
CRISPR Library construction
A list of genes that (1) encode for proteins that are targeted by small molecules in the Tchem or Tclin databases (see Pharos database [https://pharos.nih.gov]) and that (2) are expressed in erythroid cells7 was generated. A single guide RNA (sgRNA) pool containing 11 sgRNAs targeting each of these genes, 11 sgRNAs targeting each of the 3 known γ-globin repressors (BCL11a, LRF, and CHD4), and 550 nontargeting controls was generated using the GPP sgRNA Designer (Broad Institute). The oligo pool was purchased from Twist Bioscience and cloned into lentiCRISPRv2 (addgene #52961) by Gibson assembly. The library construction, plasmid expansion, and lentiviral packaging were done essentially as previously described.8
HuDEP2 transduction
HuDEP2 cells were spinfected with lentivirus with the 4 μg/mL polybrene (1000g for 2 hours at 33°C), to achieve a multiplicity of infection of 0.4. After spinfection, cells were pelleted and resuspended in fresh HuDEP2 expansion medium. Twenty-four hours later, 0.3 μg/mL of puromycin was added to the medium for 6 days to remove uninfected cells. Following puromycin selection, erythroid differentiation was performed as described in the following sections. At day 6 of differentiation, cells with the top or bottom 10% HbF expression were sorted. sgRNA sequences were amplified and analyzed as previously described.9
HuDEP2 cell culture and erythroid differentiation
HbF staining
HbF staining was modified from a previous publication.13 The details are listed in the supplemental Methods.
Messenger RNA analysis
Messenger RNA (mRNA) purification, complementary DNA synthesis, and quantitative reverse transcriptase-polymerase chain reaction analyses were as described previously.11
HuDEP2 gene mutation using CRISPR
Deletion of select genes was performed using the LentiCRISPR v2 vector as previously described.10 Details of protocols are available in the supplemental Methods.
Results and discussion
Generation of a custom CRISPR knockout library
To define the potentially druggable erythroid genome, we exploited a National Institutes of Health dataset (Pharos) that links the availability of small molecule protein ligands (primarily inhibitors) to disease with their phenotypic associations.14 Human genes in this database are categorized according to their target developmental level. We focused only on Tclin and Tchem to accumulate a list of genes that encode proteins that are targeted by either US Food and Drug Administration-approved drugs or by ligands that bind to them with high potency. Of the 2211 genes found in Tclin or Tchem, 1006 are expressed in human erythroid cells.7 We generated a pooled library of sgRNAs targeting these 1006 genes (11 sgRNA/gene) and as positive controls, we included 11 sgRNAs targeting each of 3 known γ-globin repressors (BCL11A, ZBTB7A, and CHD4).5 A total of 550 nontargeting sgRNAs were included as negative controls.
This sgRNA pool was cloned into a LentiCRISPRv2 backbone, packaged into virus,8 and then infected into HuDEP2 cells at a multiplicity of infection of 0.4. After puromycin selection, infected HuDEP2 cells were cultured under erythroid differentiation conditions for an additional 6 days.10 At the end of this period, cells were subjected to intracellular HbF staining followed by flow cytometry.13 The highest and lowest 10% HbF-expressing cells were sorted into pools. sgRNA enrichment was compared between the top and the bottom 10% HbF cells (analysis 1) or between the top 10% HbF cells and all cells (analysis 2) (supplemental Figure 1A).
The top 20 genes for which sgRNAs were enriched in the HbF high cells were examined further. These genes included 2 of the positive controls (BCL11A and ZBTB7A) as well as other genes previously associated with HbF repression (MBD2, DNMT1, EHMT1/2, EZH2/EED, and HDAC2).5,15-17 Notably, the strategy applied in this report resulted in the identification of 8 novel, potentially druggable, proteins that appear to repress HbF expression (supplemental Figure 1B).
To validate the novel candidates, we deleted the genes encoding each of these proteins in HuDEP2 cells and analyzed the F-cell percentage by intracellular staining and flow cytometry. Two BCL11A-targeting sgRNAs were included as positive controls and a random sequence sgRNA (nontargeting-4) was used as a negative control. Notably, of 8 novel candidate HbF repressors validated, deletion of 7 of them resulted in a significantly increased percentage of F cells (supplemental Figure 2A-B). A subset of the most robustly responsive genes was characterized further.
The impact of VHL deletion on HbF induction is HIF1α dependent
VHL is an E3 ubiquitin ligase that degrades hydroxylated HIF1α during normoxia.18 Pharmacological activation of the HIF pathway using prolyl hydroxylase inhibitors has been previously reported to induce HbF.19 In contrast, patients harboring a homozygous VHL mutation (598C>T) do not exhibit increased HbF, despite increased serum EPO level, a result of HIF pathway activation.20 Here, we wished to clarify whether VHL plays a role in HbF repression and if the latter role is mediated via the HIF signaling pathway.
To address this issue, we generated VHL-mutant HuDEP2 cells using 2 sgRNAs (VHLg1 and VHLg4). Deletion of VHL using either sgRNA significantly increased the F-cell percentage (Figure 1A-B). We next generated HuDEP2 cells that are double deleted for VHL and HIF1α (VHL−/−:HIF1α−/−). Deletion of HIF1α fully rescued the induction of F cells caused by VHL deletion (Figure 1A-B). Consistent with the flow cytometry data, VHL deletion in HuDEP2 cells results in increased γ-globin mRNA level by four to five fold, a finding that was rescued by concurrent HIF1α deletion (Figure 1C). Notably, VHL mRNA was reduced by ∼50% in cells transduced with a VHL-targeting sgRNA, whereas HIF1α mRNA was reduced by ∼60% in cells transduced with a HIF1α-targeting sgRNA (Figure 1C), confirming efficiency of the sgRNAs used in this study. Taken together, these data show that VHL represses fetal γ-globin expression by suppressing the HIF1α pathway.
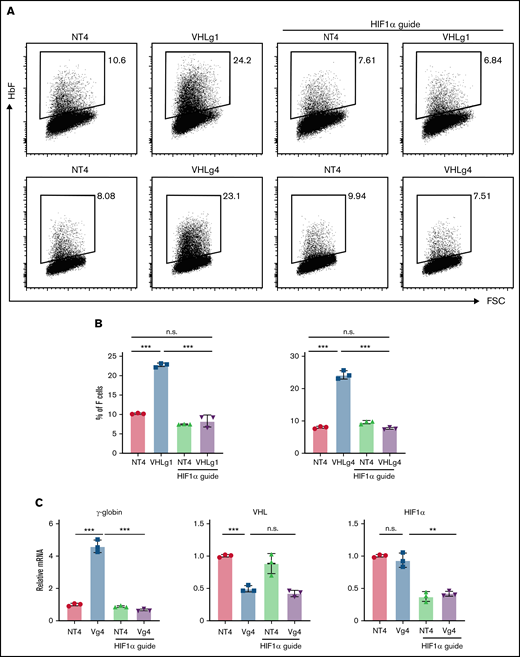
VHL deletion induces γ-globin through the activation of HIF1α. (A-B) Deletion of VHL in HuDEP2 cells using sgRNA-1 (g1) or sgRNA-4 (g4) induces HbF, a finding that is abolished by concomitant deletion of HIF1α. (C) γ-globin, VHL, and HIF1α mRNA levels were analyzed by quantitative reverse transcriptase-polymerase chain reaction. Data are shown as means ± standard deviation from 3 experiments (**P < .01; ***P < .001; n.s., not significant; unpaired Student t test).
VHL deletion induces γ-globin through the activation of HIF1α. (A-B) Deletion of VHL in HuDEP2 cells using sgRNA-1 (g1) or sgRNA-4 (g4) induces HbF, a finding that is abolished by concomitant deletion of HIF1α. (C) γ-globin, VHL, and HIF1α mRNA levels were analyzed by quantitative reverse transcriptase-polymerase chain reaction. Data are shown as means ± standard deviation from 3 experiments (**P < .01; ***P < .001; n.s., not significant; unpaired Student t test).
Notably, there is a discrepancy between no HbF induction in VHL homozygous mutant patients20 and significant HbF induction in VHL genetic knockout HuDEP2 cells. To explain this, we hypothesize that the mutant VHL protein from the 598C>T patients may still retain part of HIF1α-degrading activity, which is enough to maintain the HIF1α at the level below the threshold to induce γ-globin expression.
PTEN deletion induces HbF through induction of AKT signaling
Because PTEN is the major negative regulator of AKT signaling pathway and this pathway is involved in numerous critical biological processes,21 we chose to further investigate the potential activity of PTEN involvement in γ-globin gene induction. For this purpose, we generated PTEN mutant HuDEP2 cells using 2 PTEN-targeting sgRNAs (PTENg1 or PTENg9). Deletion of PTEN by either sgRNAs resulted in a significant increase in the percentage of F cells and the effect was rescued by cotreatment of the PTEN-mutant cells with an AKT inhibitor (supplemental Figure 3A-B). These results demonstrated that deletion of PTEN induces HbF through activation of the AKT pathway.
We next generated 2 clonal HuDEP2 cell lines with biallelic frameshift mutations in PTEN (PTEN−/− #35 and #54). Although PTEN-deficient clonal HuDEP2 cells exhibit normal erythroid differentiation (supplemental Figure 4), the percentage of F cells was significantly higher in PTEN deleted compared with control cells (Figure 2A), as was γ-globin expression at the mRNA level (Figure 2B).

Deletion and pharmacological inhibition of PTEN induces γ-globin. (A) HbF induction in 2 independent PTEN-deficient clonal HuDEP2 cell lines (PTEN−/− #35 and #54) compared with control cells (nontargeting-4), as demonstrated by HbF staining and flow cytometry (n = 3). (B) Analysis of γ-globin and β-globin mRNA levels in PTEN-deleted clonal cells (n = 3). (C-F) CD34+ cells isolated from healthy donors were differentiated into erythroid cells and treated with LSD1 (GSK690), EZH (Taze), or PTEN inhibitors (bpV), showing induction of HbF with all 3 compounds, as demonstrated by HbF staining and flow cytometry (C-D; n = 5) and mRNA analysis (E-F; n = 4). (G-H) CD34+ cells isolated from β-thalassemia patients were differentiated into erythroid cells and treated with EZH (Taze, n = 3) or PTEN (bpV, n = 6) inhibitors. Both compounds result in HbF induction by high-performance liquid chromatography analysis. Data are shown are means ± standard deviation (A-B,E-F: **P < .01; ***P < .001; n.s., not significant; unpaired Student t test; D,H: *P < .05; **P < .01; paired Student t test).
Deletion and pharmacological inhibition of PTEN induces γ-globin. (A) HbF induction in 2 independent PTEN-deficient clonal HuDEP2 cell lines (PTEN−/− #35 and #54) compared with control cells (nontargeting-4), as demonstrated by HbF staining and flow cytometry (n = 3). (B) Analysis of γ-globin and β-globin mRNA levels in PTEN-deleted clonal cells (n = 3). (C-F) CD34+ cells isolated from healthy donors were differentiated into erythroid cells and treated with LSD1 (GSK690), EZH (Taze), or PTEN inhibitors (bpV), showing induction of HbF with all 3 compounds, as demonstrated by HbF staining and flow cytometry (C-D; n = 5) and mRNA analysis (E-F; n = 4). (G-H) CD34+ cells isolated from β-thalassemia patients were differentiated into erythroid cells and treated with EZH (Taze, n = 3) or PTEN (bpV, n = 6) inhibitors. Both compounds result in HbF induction by high-performance liquid chromatography analysis. Data are shown are means ± standard deviation (A-B,E-F: **P < .01; ***P < .001; n.s., not significant; unpaired Student t test; D,H: *P < .05; **P < .01; paired Student t test).
EZH and PTEN inhibitors induce HbF in human CD34+ cells from both healthy donors and β-thalassemia patients
Finally, we asked whether pharmacological inhibition of PTEN could induce HbF under erythroid differentiation conditions in human CD34+ hematopoietic stem and progenitor cells.10 In these experiments, the LSD1 inhibitor GSK690 was used as a positive control for HbF induction.10 Because EZH2 was previously identified as a candidate γ-globin corepressor17 (a finding corroborated in our screen; supplemental Figure 1B), and because there are no published data showing that pharmacological inhibition of EZH activity induces HbF, we tested whether a clinically approved EZH inhibitor (Tazemetostat [Taze]) also results in increased HbF production. Both the PTEN inhibitor (bpV) and the EZH inhibitor (Taze) significantly induced HbF as demonstrated by HbF staining (Figure 2C-D), a finding that appears to be mediated at the transcriptional level (Figure 2E-F) in CD34+ cells derived from multiple healthy donors. Next, we investigated the efficacy of PTEN and EZH inhibitors on HbF induction in CD34+ cells recovered from multiple HbE/β-thalassemia patients. Taze and bpV significantly induced HbF by 22% and 7.5%, respectively (Figure 2G-H).
In summary, we identified novel genes that regulate γ-globin expression and showed that EZH and PTEN inhibitors significantly induce HbF. These studies support potential future development of more potent inhibitors of novel γ-globin repressors as novel therapeutics for the treatment of patients with β-globinopathies.
Authorship
Contribution: L.Y., G.M., N.J., and J.D.E. designed research; L.Y., G.M., E.S., Y.W., R.M., D.S., V.T., G.B.-C., and P.P. performed research; G.M. and R.K. contributed vital new reagents; and L.Y., G.M., K.C.L., D.G., S.A.S., N.J., and J.D.E. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James Douglas Engel, Department of Cell and Developmental Biology, 3035 BSRB, 109 Zine Pitcher Pl, Ann Arbor, MI 48109-2200; e-mail: engel@umich.edu.

