

Key Points
Activating maternal KIR-fetal HLA interactions decrease the risk of childhood acute lymphoblastic leukemia in Latinos.
Immune receptor interactions are associated with cytokine levels in Latinos and non-Latino Asians.

Abstract
Acute lymphoblastic leukemia (ALL) in children is associated with a distinct neonatal cytokine profile. The basis of this neonatal immune phenotype is unknown but potentially related to maternal-fetal immune receptor interactions. We conducted a case-control study of 226 case child-mother pairs and 404 control child-mother pairs to evaluate the role of interaction between HLA genotypes in the offspring and maternal killer immunoglobulin-like receptor (KIR) genotypes in the etiology of childhood ALL, while considering potential mediation by neonatal cytokines and the immune-modulating enzyme arginase-II (ARG-II). We observed different associations between offspring HLA-maternal KIR activating profiles and the risk of ALL in different predicted genetic ancestry groups. For instance, in Latino subjects who experience the highest risk of childhood leukemia, activating profiles were significantly associated with a lower risk of childhood ALL (odds ratio [OR] = 0.59; 95% confidence interval [CI], 0.49-0.71) and a higher level of ARG-II at birth (coefficient = 0.13; 95% CI, 0.04-0.22). HLA-KIR activating profiles were also associated with a lower risk of ALL in non-Latino Asians (OR = 0.63; 95% CI, 0.38-1.01), although they had a lower tumor necrosis factor-α level (coefficient = −0.27; 95% CI, −0.49 to −0.06). Among non-Latino White subjects, no significant association was observed between offspring HLA-maternal KIR interaction and ALL risk or cytokine levels. The current study reports the association between offspring HLA-maternal KIR interaction and the development of childhood ALL with variation by predicted genetic ancestry. We also observed some associations between activating profiles and immune factors related to cytokine control; however, cytokines did not demonstrate causal mediation of the activating profiles on ALL risk.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common type of cancer diagnosed among children under 15 years of age.1 The risk of childhood ALL varies across race/ethnic groups, with the highest incidence rate in the United States among Latino children.2 Patterns of early-life infections,3 exposure to infectious agents via childhood contact (eg, daycare attendance4 or older siblings5 ), and immunizations6 may affect the risk of childhood ALL.7 Notably, exposure to extensive social contacts and microbial stimuli in early life is protective for childhood ALL, possibly by facilitating development of a well-modulated immune system that appropriately reacts to infections.5,8 A child whose immune system was naïve to antigenic stimuli may respond to common infections in an aberrantly strong manner, stimulating B-cell growth and increasing the risk of ALL.9,10 Incorporating observations on responses to infections and the history and timing of ALL mutations, Greaves and Wiemels11 proposed that ALL evolves in 2 discrete steps. First, a preleukemic clone is generated in utero by fusion gene formation or an aneuploidy event. Then, the preleukemic clones convert to leukemia because of secondary genetic changes after birth that are driven by vigorous immune activation via common infections.5,12,13 This hypothesis proposes a role for infection in early life but does not address the immune responder status at birth before exposure to infectious agents.
More recently, studies examining neonatal cytokine levels suggest that immune status at birth plays a key role in the development of childhood ALL. Immunologically naïve newborns who had a deficit of interleukin 10 (IL-10) were at a higher risk for ALL later in childhood.14 IL-10 is a cytokine that limits the magnitude of immune response to pathogens and during pregnancy helps ensure the coexistence of fetal tissues within the mother despite the expression of “non-self” antigens from the father.15 Two other studies confirmed and extended these observations to additional cytokines, suggesting that a cluster of cytokines is crucial.16,17 The origin of this cytokine profile associated with ALL risk, along with its role in establishing the “responder status” of the infant, would provide potential avenues for ALL prevention.
The presence of specific activating killer immunoglobulin-like receptor (KIR) genes in the child is associated with a decreased risk of ALL.18,19 KIRs expressed on natural killer (NK) cells help the host to identify normal cells by the HLA expressed on the surface of these cells. HLA expression is often lost during neoplastic transformation. Cells without class I HLA expression will be targeted for removal.20 HLA and KIR expression are also fundamental in the context of normal pregnancy. Because the fetus is a foreign tissue in the mother’s uterus, the interaction between HLA and KIR is crucial in modulating maternal immune response to the developing fetus, which expresses specific HLA alleles that communicate through maternal KIR receptors to help modulate maternal NK-cell activity.21 Furthermore, it is hypothesized that during uterine NK-cell development, the maternal KIR interacts with her own HLA molecules; thus, her NK cells are “licensed” not to target the fetus’ cells with the same type of HLA molecules during placentation.22-25
Twelve KIR genes and 2 pseudogenes are classified to 2 haplotypes A and B. Haplotype A consists of 5 genes (KIR2DL1, 2DL3, 3DL1, 3DL2, 3DL3) that inhibit, 1 gene (KIR2DS4) that activates, and 1 gene (KIR2DL4) that may activate or inhibit NK-cell activity. KIR haplotype B has variable gene contents and 1 or more of the B-specific genes or alleles (KIR2DS1, 2DS2, 2DS3, 2DS5, 2DL2, 2DL5, 3DS1).26 During pregnancy, specific maternal KIRs interact with specific offspring HLA-C, -B, and -G to regulate immune responses; these interactions are classified as activating or inhibiting and form the basis for education of uterine NK cells.20,27 Our hypothesis is that, on balance, stronger interactions that activate NK-cell activity will lead to a greater degree of immunosuppression28 (higher IL-10 levels for instance14 ) and a relatively quiescent neonatal immune system reflected by a characteristic cytokine profile. This will result in lower leukemia risk as the neonate will react more moderately on exposure to infectious agents and antigens after birth.14,29
In the current study, we leveraged unique resources to evaluate whether interaction between specific maternal KIR and offspring HLA genotypes is associated with the risk of childhood ALL and if this association is mediated by cytokine levels at birth in various racial/ethnic groups that experience varying risks of childhood leukemia. This study provides novel insights into the role of in utero immune development in the pathogenesis of childhood ALL.
Methods
Study population
Cases were children diagnosed with ALL at the age of 0 to 14 years and included in the California Childhood Leukemia Study (CCLS) or the California Childhood Cancer Records Linkage Project (CCRLP). The details of CCLS and CCRLP have been described elsewhere.3,30,31 Briefly, both are case-control studies of childhood leukemia, where the cases were children 0 to 14 years of age at the time of leukemia diagnosis ascertained from pediatric hospitals in California (CCLS, 1995-2015) or from a linkage between statewide birth records and cancer diagnosis information from the California Cancer Registry (CCRLP, 1988-2011) and nonoverlapping population with CCLS. Control subjects for both studies were selected from California birth records and matched to cases on date of birth, sex, self-reported race/ethnicity, and derived from the same study region. CCLS cases were actively recruited, whereas CCRLP cases and controls were registry based only. Newborn blood samples for the offspring cases and controls in both studies were obtained from the California Biobank Program,32 with DNA isolated from newborn dried bloodspots Qiagen. In the CCLS, DNA for mothers was derived from buccal cell swabs, or saliva samples collected at the time of in-home personal interview. In the CCRLP, we extracted DNA from maternal blood specimens from 15 to 20 weeks of gestation with the case or control child, which were archived in 5 Southern California counties (San Diego, Imperial, Orange, Riverside, and San Bernardino) in the years 1999 to 2009. The study was approved by the California Health and Human Services Institutional Review Board (#2018-119) and conducted according to the Declaration of Helsinki.
The current analysis was restricted to CCLS and CCRLP cases and controls who had maternal DNA available. Cases with Down syndrome were excluded because of the potential confounding effects of trisomy 21 on immune development. A total of 226 case child-mother pairs and 404 control child-mother pairs were included in this analysis. The genetic ancestry of each subject was inferred by principal component (PC) analysis with subjects in 1000 Genomes project.33 A total of 2504 subjects (661 African American, 347 Latino, 504 East Asian, 503 European, and 489 South Asian) from 1000 Genomes were included as reference for the PC analysis.33 We first used Plink 2.034 to merge the variants of our study participants with the variants of the subjects in the 1000 Genomes project and performed a PC analysis. We then used Plink 2.0 to estimate the genetic ancestry of our study participants by comparing their first 2 PCs to the first 2 PCs of the reference subjects in the 1000 Genomes project whose genetic ancestry are known.35 We also plotted the first 2 PCs of the study participants to those of the 1000 Genomes reference subjects with R 4.1.136 (supplemental Figure 1).
Genotyping/imputing HLA alleles and KIR haplotypes
All cases, controls, and mothers were genotyped with single nucleotide polymorphism Thermo Fisher Latin (LAT) Axiom Array (CCRLP subjects, n = 1060) or by a custom Fluidigm HLA-KIR sequencing platform37,38 at Children’s Hospital Oakland Research Institute (CCLS subjects, n = 200).
HLA-A, -Bw4, and -C genotypes were imputed with “HLA genotype imputation with attribute bagging” (HIBAG)39 from the LAT array data (HIBAG: CCRLP subjects, n = 1060) or analyzed from direct sequencing (Fluidigm: CCLS subjects, n = 200; supplemental Figure 2). Ancestral group-specific models included in HIBAG were used for HLA imputation for predicted Latino, non-Latino White (NLW), non-Latino Black (NLB), and non-Latino Asian (NLA) subjects. The average prediction accuracy of HIBAG was 89% (European ancestry, 89%; African ancestry, 91%; Latino ancestry, 79%; other ancestry, 86%) as validated in a multiracial population.40 All mothers have the same predicted genetic ancestry as their offspring. KIR*Imp41 was used to impute KIR genotypes from LAT array data (KIR*Imp: CCRLP subjects, n = 1060) and direct sequencing (Fluidigm: CCLS subjects, n = 200; supplemental Figure 2). We reported the average predicted probability of each imputed HLA allele or KIR haplotype generated by HIBAG or KIR*Imp as an indicator for imputation quality (supplemental Table 1).
Quantifying the offspring HLA and maternal KIR interaction
We first used the broad category of KIR A/B haplotype as an indicator for the inhibition/activation of immune responses. More copies of the KIR A haplotype indicates more overall inhibition.19 In addition, we calculated an activation score and an inhibition score for different combinations of specific offspring HLA and maternal KIR genes, referred to as HLA-KIR interaction (Table 1) based on the compiled information on specific allelic combinations derived from previous studies.42,43 We assigned an activation/inhibition score 1 to the combinations that were reported to have a strong activation/inhibition or an activation/inhibition score 0.5 to the combinations that were reported to have a weak activation/inhibition in the reference studies. The activation score is a sum of all activating combinations, and the inhibition score is the sum of all inhibitory combinations within the mother/fetal dyad. Furthermore, to evaluate the HLA-KIR interaction and ALL risk considering NK licensing, we computed a licensed activation score and a licensed inhibition score based on the combination of maternal HLA, maternal KIR, and offspring HLA genes (supplemental Table 2).
Activation/inhibition effect for different combinations of maternal KIR and offspring HLA genes
| Maternal KIR gene* | Offspring HLA allele† | Inhibition score‡ | Activation score‡ |
|---|---|---|---|
| KIR2DL1 | C2 | 1 | |
| KIR2DL2 | C1 | 1 | |
| KIR2DL3 | C1 | 1 | |
| KIR2DL2 | C2 | 0.5 | |
| KIR2DL3 | C2 | 0.5 | |
| KIR3DL1 | BW4*80I | 1 | |
| KIR3DL1 | BW4*80T | 0.5 | |
| KIR3DL1 | A*23:01 | 0.5 | |
| KIR3DL1 | A*24:02 | 0.5 | |
| KIR3DL1 | A*24:03 | 0.5 | |
| KIR3DL1 | A*25:01 | 0.5 | |
| KIR3DL1 | A*32:01 | 1 | |
| KIR2DS1 | C2 | 1 | |
| KIR2DS2 | C1 | 1 | |
| KIR2DS4 | C*02:02 | 1 | |
| KIR2DS4 | C*04:01 | 1 | |
| KIR2DS4 | C*05:01 | 1 | |
| KIR2DS4 | C*01:02 | 1 | |
| KIR2DS4 | C*14:02 | 1 | |
| KIR2DS4 | C*16:01 | 1 | |
| KIR2DS5 | C2 | 1 | |
| KIR3DS1 | BW4*80I | 1 |
| Maternal KIR gene* | Offspring HLA allele† | Inhibition score‡ | Activation score‡ |
|---|---|---|---|
| KIR2DL1 | C2 | 1 | |
| KIR2DL2 | C1 | 1 | |
| KIR2DL3 | C1 | 1 | |
| KIR2DL2 | C2 | 0.5 | |
| KIR2DL3 | C2 | 0.5 | |
| KIR3DL1 | BW4*80I | 1 | |
| KIR3DL1 | BW4*80T | 0.5 | |
| KIR3DL1 | A*23:01 | 0.5 | |
| KIR3DL1 | A*24:02 | 0.5 | |
| KIR3DL1 | A*24:03 | 0.5 | |
| KIR3DL1 | A*25:01 | 0.5 | |
| KIR3DL1 | A*32:01 | 1 | |
| KIR2DS1 | C2 | 1 | |
| KIR2DS2 | C1 | 1 | |
| KIR2DS4 | C*02:02 | 1 | |
| KIR2DS4 | C*04:01 | 1 | |
| KIR2DS4 | C*05:01 | 1 | |
| KIR2DS4 | C*01:02 | 1 | |
| KIR2DS4 | C*14:02 | 1 | |
| KIR2DS4 | C*16:01 | 1 | |
| KIR2DS5 | C2 | 1 | |
| KIR3DS1 | BW4*80I | 1 |
KIR imputed with KIR*Imp or directly genotyped (CCLS samples only).
HLA imputed with HIBAG by predicted genetic ancestry group or directly genotyped (CCLS samples only).
Inhibition/activation score = 1 if indicated as strong inhibition/activation in previous study; score = 0.5 if indicated as weak inhibition/activation in previous study.
Cytokines
The neonatal levels of 10 cytokines (IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, interferon-γ, tumor necrosis factor α [TNF-α], and vascular endothelial growth factor) and the immune-modulating enzyme arginase-II (ARG-II) were assessed by Luminex technology on blood spot extracts or enzyme-linked immunosorbent assay, as previously described.17,44 The cytokines were normalized with a variance stabilizing normalization (VSN) method, normalizing on case/control status, birth year, protein, batch, and plate spot.45
Statistical analysis
A potential association between maternal/fetal HLA-KIR interaction and the risk of childhood ALL was analyzed with logistic regression, without matching between cases and controls, and stratified by predicted genetic ancestry (Latino/NLW/NLB/NLA) to account for potential effect modification by ancestry among the broad four groups; this was the main analysis of interest. The dependent variable of the logistic regression model was ALL case/control status, and the independent variables were the inhibition and activation scores of HLA-KIR interaction as described above. We also performed analyses adjusting for the top 5 PCs of predicted genetic ancestry to minimize the residual confounding within each ancestral group. We then used conditional logistic regression to perform a matched analysis as a sensitivity analysis to control for the potential confounding by offspring sex and genetic ancestral components. Similar to the main analyses, we built models stratified by predicted genetic ancestry (Latino/NLW/NLB/NLA). For each model, the activation and inhibition scores are the main independent variables and predicted genetic ancestry (5 PCs) is a covariate for the matched analyses. Finally, for both the main and sensitivity analyses, we used a random-effects meta-analysis model, weighted by the number of subjects in each predicted genetic ancestry group, to obtain a weighted average odds ratio (supplemental Figure 2). In addition, we used an unmatched logistic regression model to evaluate the HLA-KIR interaction and ALL risk considering NK licensing. The dependent variable is ALL case/control status, and the independent variables are licensed activation score and licensed inhibition score.
We performed an unmatched analysis to assess the relation of normalized neonatal levels of the 10 cytokines and ARG-II (dependent variable) to HLA-KIR interaction (independent variable) with a linear regression model adjusting for age at bloodspot collection, birth weight, and gestation week. The covariates were selected from a set of candidate factors (offspring sex, age at bloodspot collection, birth weight, gestation week, and type of health insurance) with a stepwise method. Variables with P < .05 were retained in the model. We built models including the activation and inhibition scores with and without adjustment for covariates. In the matched analyses, we used a linear mixed model adjusting for the same set of covariates as the linear regression model, and cluster by matched pairs, to analyze the association between the HLA-KIR scores and normalized cytokine or ARG-II levels (supplemental Figure 2).
To evaluate the potential mediation effect of cytokines and ARG-II in the association between HLA-KIR interaction and childhood ALL risk, a causal mediation analysis was performed when (1) the association between HLA-KIR interaction and childhood ALL risk was statistically significant; and (2) the association between the potential mediator (cytokine/ARG-II) and HLA-KIR interaction was statistically significant. We first evaluated the effect of the potential mediator on childhood ALL risk with a logistic regression model where the case/control status of childhood ALL was the dependent variable, and the potential mediator and HLA-KIR activation or inhibition scores were the independent variables. We then performed the causal mediation analysis.46 Briefly, the unstandardized indirect effects for each of 1000 bootstrapped samples were computed, and the 95% confidence intervals (CIs) were computed by determining the indirect effects at the 2.5th and 97.5th percentiles.
Our activation score was also tested for association with recurrent fetal loss, another outcome previously associated with maternal KIR-child HLA interactions.47 We computed activation and inhibition scores for mothers who experienced prior fetal loss (ever) with those who did not (never). Distribution by case/control status was analyzed with a χ2 test for categorical variables and 2-sample t test for continuous variables.
All statistical tests were 2-sided, and P < .05 was considered statistically significant.
Results
Offspring HLA-maternal KIR interaction and ALL risk
For the primary (unmatched) analysis, 390 (61.9%) Latino, 160 (25.4%) NLW, 64 NLA (10.2%), and 16 (2.5%) NLB child-mother pairs were included in each model (Table 2). For the sensitivity (matched) analysis, 139 Latino (62.6%), 56 NLW (25.2%), and 27 NLA (12.2%) 1-to-1 matched mother-child case-control pairs from CCLS and CCRLP were included in each model (supplemental Table 3). The distribution of maternal KIR genes and offspring HLA-C alleles are reported in supplemental Tables 4 and 5.
Demographics, offspring HLA-C, and maternal KIR carrier frequencies among case and control mother-child pairs in CCRLP and CCLS
| Latino all races | Non-Latino White | Non-Latino Asian | Non-Latino Black | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case (N = 139) | Control (N = 251) | P* | Case (N = 56) | Control (N = 104) | P* | Case (N = 27) | Control (N = 37) | P* | Case (N = 4) | Control (N = 12) | P* | |
| Study† | NA | NA | NA | NA | ||||||||
| CCRLP | 76 (54.7%) | 251 (100%) | 38 (67.9%) | 104 (100%) | 12 (44.4%) | 37 (100%) | 0 (0%) | 12 (100%) | ||||
| CCLS | 63 (45.3%) | 0 (0%) | 18 (32.1%) | 0 (0%) | 15 (55.6%) | 0 (0%) | 4 (100%) | 0 (0%) | ||||
| Offspring sex | .449 | .165 | .396 | 1.000 | ||||||||
| Female | 53 (38.1%) | 107 (42.6%) | 27 (48.2%) | 37 (35.6%) | 8 (29.6%) | 16 (43.2%) | 1 (25.0%) | 3 (25.0%) | ||||
| Male | 86 (61.9%) | 144 (57.4%) | 29 (51.8%) | 67 (64.4%) | 19 (70.4%) | 21 (56.8%) | 3 (75.0%) | 9 (75.0%) | ||||
| Offspring HLAC | .128 | .371 | .067 | .334 | ||||||||
| C1/C1 | 47 (33.8%) | 101 (40.2%) | 16 (28.6%) | 41 (39.4%) | 10 (37.0%) | 15 (40.5%) | 2 (50.0%) | 3 (25.0%) | ||||
| C1/C2 | 60 (43.2%) | 112 (44.6%) | 31 (55.4%) | 47 (45.2%) | 9 (33.3%) | 19 (51.4%) | 1 (25.0%) | 8 (66.7%) | ||||
| C2/C2 | 32 (23.0%) | 38 (15.1%) | 9 (16.1%) | 16 (15.4%) | 8 (29.6%) | 3 (8.1%) | 1 (25.0%) | 1 (8.3%) | ||||
| Maternal KIR | .002 | .939 | .139 | .120 | ||||||||
| A/A | 22 (15.8%) | 14 (5.6%) | 6 (10.7%) | 13 (12.5%) | 3 (11.1%) | 0 (0%) | 3 (75.0%) | 2 (16.7%) | ||||
| B/x | 117 (84.2%) | 237 (94.4%) | 50 (89.3%) | 91 (87.5%) | 24 (88.9%) | 37 (100%) | 1 (25.0%) | 10 (83.3%) | ||||
| Activation score | <.001 | .981 | .040 | <.001 | ||||||||
| Mean (SD) | 1.58 (1.34) | 2.44 (1.26) | 2.05 (1.52) | 2.05 (1.12) | 1.89 (1.45) | 2.62 (1.26) | 0 (0) | 1.92 (1.38) | ||||
| Inhibition score | .003 | .727 | .566 | .027 | ||||||||
| Mean (SD) | 3.41 (0.996) | 3.73 (1.03) | 3.84 (1.15) | 3.90 (1.05) | 3.52 (1.03) | 3.68 (1.13) | 2.88 (0.854) | 4.38 (1.09) | ||||
| Latino all races | Non-Latino White | Non-Latino Asian | Non-Latino Black | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case (N = 139) | Control (N = 251) | P* | Case (N = 56) | Control (N = 104) | P* | Case (N = 27) | Control (N = 37) | P* | Case (N = 4) | Control (N = 12) | P* | |
| Study† | NA | NA | NA | NA | ||||||||
| CCRLP | 76 (54.7%) | 251 (100%) | 38 (67.9%) | 104 (100%) | 12 (44.4%) | 37 (100%) | 0 (0%) | 12 (100%) | ||||
| CCLS | 63 (45.3%) | 0 (0%) | 18 (32.1%) | 0 (0%) | 15 (55.6%) | 0 (0%) | 4 (100%) | 0 (0%) | ||||
| Offspring sex | .449 | .165 | .396 | 1.000 | ||||||||
| Female | 53 (38.1%) | 107 (42.6%) | 27 (48.2%) | 37 (35.6%) | 8 (29.6%) | 16 (43.2%) | 1 (25.0%) | 3 (25.0%) | ||||
| Male | 86 (61.9%) | 144 (57.4%) | 29 (51.8%) | 67 (64.4%) | 19 (70.4%) | 21 (56.8%) | 3 (75.0%) | 9 (75.0%) | ||||
| Offspring HLAC | .128 | .371 | .067 | .334 | ||||||||
| C1/C1 | 47 (33.8%) | 101 (40.2%) | 16 (28.6%) | 41 (39.4%) | 10 (37.0%) | 15 (40.5%) | 2 (50.0%) | 3 (25.0%) | ||||
| C1/C2 | 60 (43.2%) | 112 (44.6%) | 31 (55.4%) | 47 (45.2%) | 9 (33.3%) | 19 (51.4%) | 1 (25.0%) | 8 (66.7%) | ||||
| C2/C2 | 32 (23.0%) | 38 (15.1%) | 9 (16.1%) | 16 (15.4%) | 8 (29.6%) | 3 (8.1%) | 1 (25.0%) | 1 (8.3%) | ||||
| Maternal KIR | .002 | .939 | .139 | .120 | ||||||||
| A/A | 22 (15.8%) | 14 (5.6%) | 6 (10.7%) | 13 (12.5%) | 3 (11.1%) | 0 (0%) | 3 (75.0%) | 2 (16.7%) | ||||
| B/x | 117 (84.2%) | 237 (94.4%) | 50 (89.3%) | 91 (87.5%) | 24 (88.9%) | 37 (100%) | 1 (25.0%) | 10 (83.3%) | ||||
| Activation score | <.001 | .981 | .040 | <.001 | ||||||||
| Mean (SD) | 1.58 (1.34) | 2.44 (1.26) | 2.05 (1.52) | 2.05 (1.12) | 1.89 (1.45) | 2.62 (1.26) | 0 (0) | 1.92 (1.38) | ||||
| Inhibition score | .003 | .727 | .566 | .027 | ||||||||
| Mean (SD) | 3.41 (0.996) | 3.73 (1.03) | 3.84 (1.15) | 3.90 (1.05) | 3.52 (1.03) | 3.68 (1.13) | 2.88 (0.854) | 4.38 (1.09) | ||||
The difference distribution by case/control status was analyzed with a χ2 test for categorical variables and 2-sample t test for continuous variables. P < .05 was considered statistically significant.
The P values were NA because there was no control in CCLS.
In our primary analysis, the activating HLA-KIR interaction was inversely associated with ALL risk in Latino subjects but not in other ancestry groups. Genetic ancestry was a significant effect modifier between ALL case/control status and the activating HLA-KIR interaction (log-likelihood P = .003). The weighted average of unadjusted odds ratios (ORs) among Latino, NLW, and NLA subjects were not different from the null for both the activation (OR = 0.69; 95% CI, 0.47-1.03) and inhibition scores (OR = 0.87; 95% CI, 0.73-1.02). In the multivariable model for Latinos, the odds of childhood ALL decreased by 41% with each 1-unit increase in activation score (OR = 0.59; 95% CI, 0.49-0.71; P < .001) when adjusting for inhibition score and the top 5 PCs for predicted genetic ancestry. The weighted average of adjusted ORs were similar to those of the unadjusted ORs (ORactivationscore = 0.69; 95% CI, 0.45-1.04; ORinhibitionscore = 0.85; 95% CI, 0.71-1.01; Table 3). Similar effect estimates in Latino subjects were observed in the univariate model (OR = 0.60; 95% CI, 0.49-0.72; P < .001), as well as with the logistic regression models where cases and controls were matched on sex (unadjusted OR = 0.68; 95% CI, 0.55-0.85; P < .001; adjusted OR = 0.66; 95% CI, 0.53-0.83; P < .001). Similar weighted average ORs and indicator of heterogeneity were observed in the matched models compared with the unmatched models (supplemental Table 6). In the analysis where maternal NK licensing with her own MHC class I–specific alleles was considered (which would happen prior to and during pregnancy), activation and inhibition scores were attenuated, and no significant association was found between ALL risk and the licensed activation or licensed inhibition scores (supplemental Table 7).
Logistic regression models assessing the association between childhood ALL case/control status and HLA-KIR interaction
| Predicted genetic ancestry | Type of score | Unadjusted model | Model adjusting for top 5 PCs | ||
|---|---|---|---|---|---|
| Odds ratio (95% CI) | P* | Odds ratio (95% CI) | P* | ||
| Latino all races | Activation score | 0.60 (0.49-0.72) | <.001 | 0.59 (0.49-0.71) | <.001 |
| Inhibition score | 0.82 (0.66-1.01) | .066 | 0.81 (0.64-1.01) | .057 | |
| Non-Latino White | Activation score | 1.02 (0.78-1.32) | .907 | 1.03 (0.78-1.35) | .842 |
| Inhibition score | 0.94 (0.69-1.29) | .705 | 0.91 (0.66-1.27) | .592 | |
| Non-Latino Asian | Activation score | 0.65 (0.42-1.00) | .051 | 0.63 (0.38-1.01) | .052 |
| Inhibition score | 0.98 (0.59-1.62) | .945 | 0.91 (0.51-1.59) | .729 | |
| P-value (LR test) | Activation score† | .003 | .003 | ||
| Inhibition score‡ | .335 | .380 | |||
| Weighted average§ | Activation score | 0.69 (0.47-1.03) | .068 | 0.69 (0.45-1.04) | .076 |
| Inhibition score | 0.87 (0.73-1.02) | .089 | 0.85 (0.71-1.01) | .064 | |
| Predicted genetic ancestry | Type of score | Unadjusted model | Model adjusting for top 5 PCs | ||
|---|---|---|---|---|---|
| Odds ratio (95% CI) | P* | Odds ratio (95% CI) | P* | ||
| Latino all races | Activation score | 0.60 (0.49-0.72) | <.001 | 0.59 (0.49-0.71) | <.001 |
| Inhibition score | 0.82 (0.66-1.01) | .066 | 0.81 (0.64-1.01) | .057 | |
| Non-Latino White | Activation score | 1.02 (0.78-1.32) | .907 | 1.03 (0.78-1.35) | .842 |
| Inhibition score | 0.94 (0.69-1.29) | .705 | 0.91 (0.66-1.27) | .592 | |
| Non-Latino Asian | Activation score | 0.65 (0.42-1.00) | .051 | 0.63 (0.38-1.01) | .052 |
| Inhibition score | 0.98 (0.59-1.62) | .945 | 0.91 (0.51-1.59) | .729 | |
| P-value (LR test) | Activation score† | .003 | .003 | ||
| Inhibition score‡ | .335 | .380 | |||
| Weighted average§ | Activation score | 0.69 (0.47-1.03) | .068 | 0.69 (0.45-1.04) | .076 |
| Inhibition score | 0.87 (0.73-1.02) | .089 | 0.85 (0.71-1.01) | .064 | |
A total of 390 Latino, 160 non-Latino White, and 64 non-Latino Asian mother-child pairs were included. LR test, log-likelihood ratio test.
P value based on a Wald test. Odds ratio statistically significantly different from the null with P < .05.
LR test comparing the null model (ALL case/control status ∼ activation score + inhibition score + genetic ancestry) to the model with an interaction term between ancestry and activation score (ALL case/control status ∼ activation score + inhibition score + genetic ancestry + activation score × genetic ancestry).
LR test comparing the null model (ALL case/control status ∼ activation score + inhibition score + genetic ancestry) to the model with an interaction term between ancestry and inhibition score (ALL case/control status ∼ activation score + inhibition score + genetic ancestry + inhibition score × genetic ancestry).
The weighted average of odds ratio among Latino all races, non-Latino White, and non-Latino Asian subjects. Calculated with a random-effects meta-analysis model where the weights are the number of subjects in each predicted genetic ancestry group.
Recurrent miscarriage was previously linked to weak HLA-KIR interactions47 and was evaluated here to help validate our data and activation index instrument. In CCLS and CCRLP, the status of previous fetal loss was known for 583 mothers (92.5%). Among them, 122 mothers (20.9%) had a fetal loss before giving birth to the index child. The activation score of HLA-KIR interaction was lower among mothers who had a fetal loss (mean = 2.08; standard deviation [SD] = 1.28) compared with those who never had a fetal loss at the time of giving birth to the child (mean = 2.25; SD = 1.32). This difference was not statistically significant (P = .22); however, the direction of association was consistent overall and among the 3 predicted genetic ancestry groups studied (supplemental Table 8).
Cytokines and offspring HLA-maternal KIR interaction
Information on cytokines and ARG-II were only available for CCRLP subjects. In total, 323 (63.3%) Latino, 139 (27.3%) NLW, and 48 NLA (9.4%) mother-child pairs were included in this analysis. NLB subjects were not included because none of the cases were NLB (supplemental Table 9).
With a multivariable linear regression model adjusting for age at bloodspot collection, birth weight, and gestation week, a 1-unit increase in the activation score was associated with a 0.13-unit (95% CI, 0.04-0.22) increase in normalized ARG-II level for Latinos. For NLAs, a 1-unit increase in activation score was associated with a 0.29-unit (95% CI, −0.56 to −0.02) decrease in normalized IL-10 level, a 0.27-unit (95% CI, −0.49 to −0.06) decrease in normalized TNF-α level; a 1-unit increase in inhibition score was associated with a 0.35-unit (95% CI, −0.65 to −0.05) decrease in normalized ARG II level (Figure 1; supplemental Table 10).
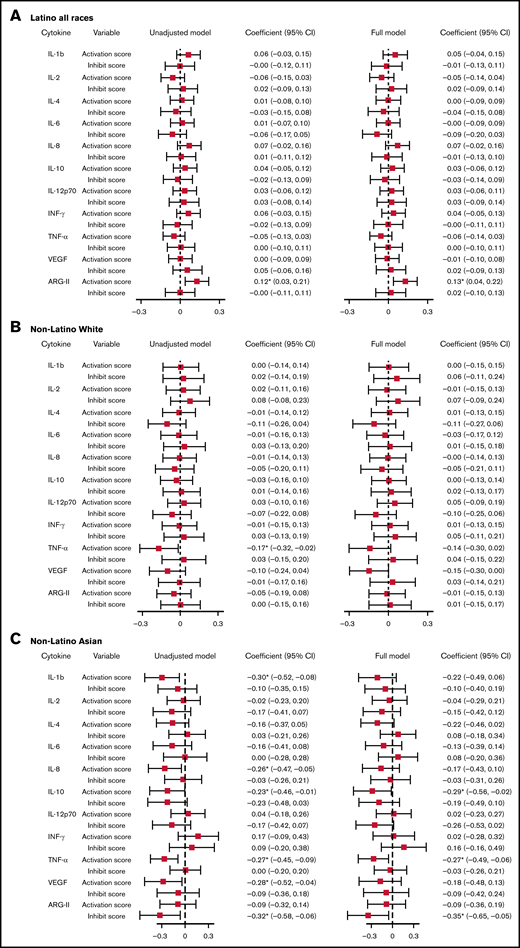
Linear regression model assessing the association between VSN normalized cytokines and activation/inhibition scores. Unadjusted model: model with cytokines as the outcome, activation, and inhibition scores as predictors. Full model: model with cytokines as the outcome, activation, and inhibition scores as predictors, adjusting for age at cytokine collection, birth weight, and gestation week. Cytokines were normalized on case/control status, birth year, protein, batch, and plate spot. *Coefficient statistically significantly different from the null with P < .05. (A) Association between VSN normalized cytokines and activation/inhibition scores among Latino subjects. (B) Association between VSN normalized cytokines and activation/inhibition scores among non-Latino White subjects. (C) Association between VSN normalized cytokines and activation/inhibition scores among non-Latino Asian subjects. INF, interferon; SE, standard error; VEGF, vascular endothelial growth factor.
Linear regression model assessing the association between VSN normalized cytokines and activation/inhibition scores. Unadjusted model: model with cytokines as the outcome, activation, and inhibition scores as predictors. Full model: model with cytokines as the outcome, activation, and inhibition scores as predictors, adjusting for age at cytokine collection, birth weight, and gestation week. Cytokines were normalized on case/control status, birth year, protein, batch, and plate spot. *Coefficient statistically significantly different from the null with P < .05. (A) Association between VSN normalized cytokines and activation/inhibition scores among Latino subjects. (B) Association between VSN normalized cytokines and activation/inhibition scores among non-Latino White subjects. (C) Association between VSN normalized cytokines and activation/inhibition scores among non-Latino Asian subjects. INF, interferon; SE, standard error; VEGF, vascular endothelial growth factor.
When matched on offspring sex and predicted genetic ancestry, 76 (60.8%) Latino, 38 (30.4%) NLW, and 11 (8.8%) NLA mother-child case-control pairs were included in the analysis (supplemental Table 11).
With a multivariable linear mixed model adjusting for age at cytokine collection, birth weight, and gestation week, we found that normalized TNF-α was the only cytokine having a statistically significant association with activation score among Latino subjects. The level of normalized TNF-α decreased by 0.15 unit (95% CI, −0.31 to −0.00) with a 1-unit increase in activation score (supplemental Table 12). We observed no associations between other cytokines or ARG-II and HLA-KIR activation score in Latino subjects or between any cytokine/ARG-II and the activation score in any other racial/ethnic groups.
To test whether cytokines were mediating the impact of HLA-KIR interactions, a mediation analysis was performed to assess the potential mediation effect of neonatal ARG-II in the association between activating HLA-KIR interaction and childhood ALL risk for Latino subjects. However, the average bootstrapped unstandardized indirect effect was 0.00214 (95% CI: −0.00404, 0.01), indicating no causal mediation (supplemental Figure 3; supplemental Table 13).
Discussion
Consistent with our original hypothesis, activating HLA-KIR interactions contributed to a lower risk of childhood ALL in Latinos and potentially subjects of other genetic ancestry groups, although the latter results did not reach statistical significance possibly because of the lack of statistical power. A weighted average meta-analysis among predicted genetic ancestry groups implied that true heterogeneity exists, which mirrors the heterogeneous findings among predicted genetic ancestry groups previously observed in California regarding immunologic risk factors such as childhood contacts exposure (daycare)48 and early child infectious disease history.3 Our finding is also compatible with a previous report that the inheritance of a higher number of activating KIR genes reduced the risk of childhood ALL in Latinos only (and not in non-Latino whites).19 Activating HLA-child/KIR-mother interactions were also protective against other conditions that can result from maladaptive maternal-fetal interactions including recurrent spontaneous abortion,49 recurrent miscarriage,50 and reproductive failure.24
In our study, activating offspring HLA-maternal KIR combinations that were associated with lower risk of childhood ALL in Latino subjects were also associated with a higher level of ARG-II, an immunosuppressive enzyme previously linked to ALL risk.44 Additionally, in NLA subjects the activating HLA-KIR combinations are associated with lower levels of IL-10 and TNF-α, and inhibiting HLA-KIR combinations are associated with lower levels of ARG-II. These findings suggest that the role of early life immune function or conditioning in the etiology of childhood ALL may vary by predicted genetic ancestry groups concerning the mediation effects of certain cytokines. There were no significant associations observed among the NLW subjects, which consisted of a substantial proportion of our study population suggesting sufficient power to see an association should one exist. In our previous work, we identified stronger associations with postnatal immune stimulatory exposures such as childhood contacts in daycare, and birth order (having older siblings) among NLW compared with Latinos,3,48 and it is possible that such postnatal exposures have a stronger impact in NLW compared with Latinos, whereas prenatal immune development by HLA-KIR interactions might play a larger role in Latinos. This is also compatible with our prior result on KIR genes only, where significant results were only apparent for Latinos in an ALL case-control study (not including mothers).19 Specifically, we reported that the frequency of KIR A/A haplotype was significantly higher in Latino cases compared with controls but not in NLW subjects. In the current study that includes a nonoverlapping sample set, we showed a consistent observation among genotyped mothers that the frequency of maternal KIR A/A was higher in Latino cases compared with controls (P = .002) but not in NLW (P = .939), NLA (P = .139), or NLB subjects (P = .120). In our previous study, we stated that external environmental factors, such as patterns of infection,51,52 that interact with KIR may help account for the variation in childhood ALL risk by ethnicity. This is a reasonable hypothesis based on the role of KIR in a nascent leukemia clone in a child after birth, where interactions with maternal alleles will not play a role. In the current study, we examined a different hypothesis impacting only the fetal period by evaluating neonatal cytokines as potential mediators of the association between child HLA/maternal KIR interaction and risk of childhood ALL. Based on the current findings, we postulate that the maternal-fetal genetic interaction between HLA (fetus) and KIR (mother) could be the root cause rather than any external environmental influence.
We found that HLA-KIR interactions are associated with ALL risk in some groups (Latinos and NLA) and that ARG-II is associated with the HLA-KIR interaction in Latino subjects; ARG-II, IL-10, and TNF-α are associated with the interaction in NLA subjects. ARG-II is a cytokine that suppresses T-cell proliferation through an anti-inflammatory cascade resulting in arginine depletion.53 Increased neonatal level of ARG-II was associated with a higher risk of childhood ALL.44 IL-10, as explained above, is a major immunosuppressive cytokine during pregnancy associated with fetal tolerance in the mother. TNF-α is an inflammatory cytokine, a component of the “cytokine storm,” and its reduced level with strong HLA-KIR interactions may be reflective of a calmed immune phenotype at birth that may affect the way an infant responds to external antigenic stimuli and infectious agents after birth.
The maternal-fetal health literature has shown that repeated miscarriages can result from weak HLA-KIR interactions.47 Our data were consistent with these prior results; although not reaching statistical significance (possibly because of limited sample size and power), we found weaker HLA-KIR interaction for mothers with a history of fetal loss compared with those without such a history among all 3 predicted genetic ancestry groups tested. Such interactions may lead to lower immunosuppression via certain cytokine profiles. Recurrent miscarriage is more frequent in mothers of children with ALL compared with controls,54 and although this was previously speculated to be a product of a higher frequency of fetal chromosome abnormalities,55,56 our results suggest that immune development may play a role.
There are several strengths and limitations of the current study. One strength is the diverse population in California, especially a large percentage of Latinos who harbor the highest risk of ALL.2 We were uniquely positioned to evaluate the association between HLA-KIR interaction and childhood ALL risk in multiple ancestral groups and did observe differences by predicted genetic ancestry. In addition, we were able to measure the level of cytokines at birth before the subjects have developed ALL by using archived newborn blood spots from the California Biobank. Furthermore, we obtained DNA from mothers, which enabled us to impute KIR genes for mothers. A limitation of the current study is that, although we imputed HLA genotypes respectively for each ethnic group, the KIR genes were imputed using European ancestry population as the reference. This may yield misclassification error in the KIR genes for subjects in minority ethnic groups. Another limitation is that the sample size of non-Latino minority Americans is small, particularly the paucity of non-Latino Black subjects, because of the composition of the California population and a lower risk of ALL in Black children. This compromises the generalizability of the results to NLB subjects. An additional limitation is that we could not adequately evaluate how NK education may have influenced the functionality of NK in stimulating cells given the extensive hypothetical assumptions of our statistical modeling leading to an algebraic treatment of the data. According to the “NK education” hypothesis, NK cells are fully responsive to target cells only when the ligand for inhibitory MHC class I–specific receptors (eg, KIR2DL1/2, KIR3DL1) are expressed in the host.57-59 NK cells lacking MHC class I–specific receptors are less responsive to target cells. Conversely, the education of NK cells via activating KIRs is reported to induce tolerance, which complements the education via inhibitory KIRs.60 However, with the current data, we could not tease out how much NK were licensed for activation or inhibition.
In summary, the current study supports the role of HLA-KIR interaction in the development of childhood ALL, with variation in effect by genetic ancestry. Further research to confirm and extend these findings is necessary, as well as the impact of postnatal infections that was not evaluated in the current study population.
Acknowledgments
The CCRLP biospecimens used in this study were obtained from the California Biobank Program (Screening Information System request number 600), in accordance with Section 6555(b), 17 CCR. The California Department of Public Health (CDPH) is not responsible for the results or conclusions drawn by the authors of this publication. The authors thank Robin Cooley and Martin Kharrazi (CDPH). For the CCLS specimens used in this work, the authors thank the families that participated in the CCLS. Without their time and effort, none of our studies would have been possible. For recruitment of subjects enrolled in the California Childhood Leukemia Study (CCLS), the authors gratefully acknowledge the clinical investigators at the following collaborating hospitals: University of California Davis Medical Center (Jonathan Ducore), University of California San Francisco (Mignon Loh and Katherine Matthay), Children’s Hospital of Central California (Vonda Crouse), Lucile Packard Children’s Hospital (Gary Dahl), Children’s Hospital Oakland (James Feusner and Carla Golden), Kaiser Permanente Roseville (formerly Sacramento) (Kent Jolly and Vincent Kiley), Kaiser Permanente Santa Clara (Carolyn Russo, Alan Wong, and Denah Taggart), Kaiser Permanente San Francisco (Kenneth Leung), Kaiser Permanente Oakland (Daniel Kronish and Stacy Month), California Pacific Medical Center (Louise Lo), Cedars-Sinai Medical Center (Fataneh Majlessipour), Children’s Hospital Los Angeles (Cecilia Fu), Children’s Hospital Orange County (Leonard Sender), Kaiser Permanente Los Angeles (Robert Cooper), Miller Children’s Hospital Long Beach (Amanda Termuhlen), University of California, San Diego Rady Children’s Hospital (William Roberts), and University of California, Los Angeles Mattel Children’s Hospital (Theodore Moore).
Research reported in this publication was supported by National Institutes Health award R01CA175737 (to J.L.W. and X.M.).
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: J.L.W., X.M., E.T., and A.J.d.S. designed research; Q.F., M.Z., S.L., and R.W. performed research; M.Z., L.M., H.H., C.M., A.K., A.L.F., D.P., H.E., and S.S.M. collected data; Q.F., J.A.H., H.E., J.L.W., and X.M. analyzed and interpreted data; Q.F. performed statistical analysis; Q.F. and J.L.W. wrote the manuscript; and J.L.W., X.M., J.A.H., A.J.d.S., N.M., M.Z., C.M., H.E., and S.S.M. edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiaomei Ma, Chronic Disease Epidemiology, PO Box 208034, 60 College St, New Haven, CT 06520; e-mail: xiaomei.ma@yale.edu; or Joseph L. Wiemels, Center for Genetic Epidemiology, 1450 Biggy St, Norris Research Tower, Room 1506A, Los Angeles, CA 90089; e-mail: wiemels@usc.edu.

