

Key Points
A reduction in hemolysis with voxelotor analog, GBT1118, reduced hemoglobinuria and kidney injury biomarkers in transgenic sickle mice.
Improved chronic hemolysis preserved kidney function and histopathologic and ultrastructural changes in transgenic sickle mice.

Introduction
Sickle cell anemia (SCA) is characterized by vaso-occlusion and chronic intravascular hemolysis, which lead to acute and chronic organ damage and early mortality. Chronic kidney disease occurs in up to half of adults with SCA1 and contributes to approximately 16% to 18% of deaths.2,3 The underlying mechanisms for the development of kidney disease are myriad and include ischemia, oxidative stress, hyperfiltration, and glomerular hypertension.4 Therapies directly targeting the pathophysiology of SCA-related kidney damage are needed.
Hemoglobin (Hb) S polymerization causes red blood cells to sickle, which leads to vaso-occlusion and hemolysis. Vaso-occlusion can promote inflammatory changes, ischemia–reperfusion injury, and vascular endothelial damage to the kidney.4 Approximately 30% of the hemolysis in SCA occurs intravascularly.5 Intravascular hemolysis results in the release of cell-free Hb, which then rapidly dissociates into dimers and is freely filtered through the glomerulus.6 Furthermore, cell-free Hb is rapidly converted to methemoglobin with the release of free heme.7 When haptoglobin and hemopexin are depleted, as occurs in SCA, cell-free Hb and heme can cause direct oxidative injury and upregulation of inflammatory, immune response, and fibrogenic pathways.7,8 The kidneys are the primary route for clearance of nonscavenged cell-free Hb and heme and are particularly susceptible to these deleterious pathways.9
Voxelotor is a small molecule allosteric Hb modulator that binds and maintains sickle Hb in the oxygenated state, thereby preventing Hb S polymerization and red blood cell sickling. In a phase 3 study of patients with SCA, voxelotor improved the degree of hemolysis, as reflected by a rise in Hb concentration and a reduction in indirect bilirubin and reticulocyte percentage.10 The benefits of reducing Hb S polymerization and hemolytic anemia with voxelotor on kidney function are unknown.
Transgenic mice harboring the Hb S mutation have been developed to investigate the pathophysiology of SCA-related complications and potential therapies to ameliorate these complications. Consistent with what has been observed in humans, sickle mice have higher concentrations of albuminuria and proteinuria compared with Hb AA mice.11,12 The transgenic sickle mice also demonstrate similar histopathologic changes with patients with SCA in the glomeruli (eg, glomerulosclerosis and membranoproliferative glomerulonephritis-like lesions), mesangium (eg, mesangial expansion and inflammatory cell infiltration), and proximal tubules (eg, increased iron deposition, atrophy, and increased basement membrane thickening).12-16 We investigated whether GBT1118, an analog of voxelotor designed to achieve similar pharmacokinetic properties in transgenic sickle mice,17 would improve biomarkers of kidney damage and kidney function in SCA.
Methods
Animal procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago (UIC). Transgenic sickle cell mice (Townes model, Jackson Laboratory; Bar Harbor, ME) were bred and housed in the UIC Biologic Resources Laboratory. Studies were conducted in age- and sex-matched Hb AA and SS mice (5 males and 5 females per group). Hb SS mice were treated with either GBT1118 chow or control chow, both provided by Global Blood Therapeutics (South San Francisco, CA), from 12 to 24 weeks of age and sacrificed after treatment.
Mice were placed in standard metabolic cages and allowed to adapt for 1 day before obtaining 24-hour urine samples at baseline and 3-week intervals for measuring cell-free Hb and biomarkers of reactive oxygen species damage (thiobarbituric acid reactive substances [TBARS]), kidney injury (KIM-1 [kidney injury molecule-1] and nephrin), and kidney dysfunction (urine albumin and protein). Blood (≤60 µL) was collected via retro-orbital bleeding into sodium EDTA tubes under isoflurane anesthesia at baseline, 6 weeks, and 12 weeks of treatment to determine GBT1118-Hb occupancy, Hb concentration, absolute reticulocyte counts, and serum cystatin C, blood urea nitrogen (BUN), and creatinine concentrations.
Excised renal cortical tissue was fixed in formalin or dissected into 1-mm sections and fixed in Trump’s fixative (EMS; Hatfield, PA), followed by processing by the UIC Research Histology Core and UIC Electron Microscopy Core, respectively. Histopathology was evaluated by a renal pathologist and an experienced researcher blinded to the treatment groups.
RNA was extracted and processed from glomeruli (isolated using the sieving method)18,19 and cortical tissue by the UIC, Core Genomics Facility. Illumina NovaSeq6000 was employed for the gene expression studies. Detection of differentially expressed genes and gene enrichment pathway analysis were performed as previously described.12
Variables were compared by treatment status with the Mann-Whitney test or ANOVA, adjusting for sex, using Systat 13 (Systat Software Corporation; Chicago, IL).
Additional methods are described in the supplemental Material.
Results and discussion
Baseline characteristics were similar between the treated vs control (untreated) sickle mice at 12 weeks of age (supplemental Table 1). Hb occupancy of GBT1118 after 6 weeks (33% ± 2%) and 12 weeks (28% ± 2%) of therapy was similar to Hb occupancy of voxelotor in the phase 3 SCA clinical study (27%).10 Also, consistent with the clinical data, sickle mice treated with GBT1118 demonstrated a reduction in hemolysis, as reflected by increases in Hb concentrations and reductions in absolute reticulocyte counts compared with control sickle mice (Figure 1A-B).

Changes in markers of hemolysis based on Hb concentration (A) and absolute reticulocyte counts in the blood (B), and cell-free Hb (C) in the urine of GBT1118 treated vs control (untreated) sickle mice. Urine biomarkers of reactive oxygen species (TBARS) (D), glomerular (nephrin) (E), and tubular injury (KIM-1) (F) in GBT1118-treated control sickle and Hb AA mice. Measures of kidney dysfunction as assessed by 24-hour urine albumin (G), 24-hour urine protein concentration (H), and serum cystatin C (I) in GBT1118-treated sickle, control sickle, and Hb AA mice. Each group contained 10 mice. *P < .05 and **P < .01 for differences between the GBT1118-treated vs control sickle mice.
Changes in markers of hemolysis based on Hb concentration (A) and absolute reticulocyte counts in the blood (B), and cell-free Hb (C) in the urine of GBT1118 treated vs control (untreated) sickle mice. Urine biomarkers of reactive oxygen species (TBARS) (D), glomerular (nephrin) (E), and tubular injury (KIM-1) (F) in GBT1118-treated control sickle and Hb AA mice. Measures of kidney dysfunction as assessed by 24-hour urine albumin (G), 24-hour urine protein concentration (H), and serum cystatin C (I) in GBT1118-treated sickle, control sickle, and Hb AA mice. Each group contained 10 mice. *P < .05 and **P < .01 for differences between the GBT1118-treated vs control sickle mice.
There were also reductions in hemoglobinuria (Figure 1C) and urine concentrations of TBARS, a biomarker of oxidative injury (Figure 1D). Furthermore, urine biomarkers of glomerular (nephrin) and proximal tubular (KIM-1) injury were improved in the GBT1118-treated vs control sickle mice (Figure 1E-F). Albuminuria and proteinuria stabilized in treated sickle mice but progressively increased in control sickle mice (Figure 1G-H). At the end of treatment, 24-week-old GBT1118-treated mice had serum cystatin C, creatinine, and BUN concentrations that were similar to 12-week-old sickle mice before treatment and to 24-week-old Hb AA mice; 24-week-old control sickle mice had significantly higher serum cystatin C, creatinine, and BUN concentrations (Figure 1I; supplemental Figure 1).
Immunofluorescence microscopy demonstrated that glomerular nephrin retention, interstitial macrophage infiltration, and collagen type IV deposition were similar between the 24-week-old GBT1118-treated and 12-week-old sickle mice (Figure 2A; supplemental Figure 2). In contrast, the 24-week-old control sickle mice had a further reduction of nephrin from the glomerulus and increased interstitial macrophage infiltration and collagen type IV deposition.
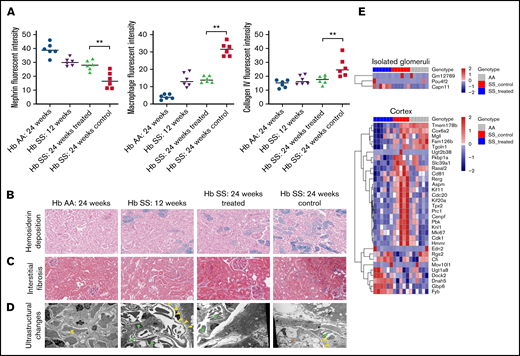
(A) Fluorescence intensity of nephrin retained in the glomerulus, interstitial macrophage infiltration (defined by F4/F80-positive cells), and collagen IV deposition in the transgenic sickle and Hb AA mice. Representative images at 200× of hemosiderin deposition by Prussian blue stain (B) and interstitial fibrosis by Masson trichrome stain (C) in the transgenic sickle and Hb AA mice. (D) Representative images of ultrastructural changes by transmission electron microscopy. Yellow arrows represent foot process effacement, green arrows represent segmental basement membrane reduplication and subendothelial electron-lucent widening, and orange asterisks represent segmental mesangial interposition with new basement membrane formation. (E) Heat map of genes differentially expressed in isolated glomeruli and kidney cortices of GBT1118-treated sickle, control sickle, and Hb AA mice (false discovery rate <0.05). **P < .01 for differences between the GBT1118-treated vs control sickle mice.
(A) Fluorescence intensity of nephrin retained in the glomerulus, interstitial macrophage infiltration (defined by F4/F80-positive cells), and collagen IV deposition in the transgenic sickle and Hb AA mice. Representative images at 200× of hemosiderin deposition by Prussian blue stain (B) and interstitial fibrosis by Masson trichrome stain (C) in the transgenic sickle and Hb AA mice. (D) Representative images of ultrastructural changes by transmission electron microscopy. Yellow arrows represent foot process effacement, green arrows represent segmental basement membrane reduplication and subendothelial electron-lucent widening, and orange asterisks represent segmental mesangial interposition with new basement membrane formation. (E) Heat map of genes differentially expressed in isolated glomeruli and kidney cortices of GBT1118-treated sickle, control sickle, and Hb AA mice (false discovery rate <0.05). **P < .01 for differences between the GBT1118-treated vs control sickle mice.
The degrees of hemosiderin deposition and interstitial fibrosis were stable in the cortex of the 24-week-old treated sickle mice but increased in 24-week-old control sickle mice compared with 12-week-old sickle mice (Figure 2B-C). Hemosiderin deposition became evident in the Bowman’s capsule of 24-week-old control sickle mice.
Transmission electron microscopy of the sickle mice demonstrated segmental foot process effacement, segmental basement membrane reduplication, and subendothelial electron-lucent widening (Figure 2D). Twelve- and 24-week-old GBT1118-treated sickle mice had predominantly intact foot processes, while 24-week-old control mice had approximately 30% podocyte foot process effacement as well as segmental areas of lobular changes with mesangial interposition interspersed by new basement membrane formation.
Three genes were differentially expressed in the glomeruli, and 32 genes in the kidney cortex of GBT1118-treated vs control sickle mice (false discovery rate <0.05) (Figure 2E; supplemental Table 2). The upregulation of Pou4f2 and Capn11 in the glomeruli of GBT1118-treated sickle mice may lead to enhanced podocyte health. Pou4f2 is coexpressed with Wt1, and its functions include maintaining podocytes in their differentiated epithelial state.20 The calpain system, including Capn11, encodes cysteine proteases that inhibit autophagy under inflammatory stimuli.21 Capn11 also facilitates the degradation of extracellular matrix and cytoskeletal remodeling and has been recently identified as a target gene located in the chronic kidney disease-associated susceptibility region of the variant, rs881858, in chromosome 6.22
In the kidney cortex, gene enrichment analysis demonstrated downregulation of several genes involved in cell cycle and mitosis, cytoskeleton, and microtubule formation in GBT1118-treated vs control sickle mice (supplemental Table 3). Genes of particular interest that were differentially expressed include upregulation of genes involved in protection against complement activation (Cfi) and angiotensin-activated signaling (Rgs2) and downregulation of genes activated under oxidative stress (Cox6a2) in the treated vs untreated sickle mice. Complement factor inhibitor (Cfi) inactivates C3b, thereby preventing overactivation of the complement alternative pathway. Impaired Cfi function has been implicated in other glomerulopathies, such as atypical hemolytic uremic syndrome and C3 glomerulonephritis.23 Dysregulated complement activation may also promote kidney damage in SCA. Increased C3 expression and C3 deposition have been described in the kidneys of sickle vs nonsickle mice12 and in patients with SCA with vs without kidney disease,24 respectively. Angiotensin II upregulates proinflammatory and fibrotic signaling pathways and has been linked to SCA and non–SCA-related kidney damage.25 Upregulation of Rgs2 in human embryonic kidney cells inhibits angiotensin II-induced profibrotic ERK activation and proinflammatory CXCR4 production.26 Furthermore, Rgs2-deficient mice demonstrate increased collagen deposition and macrophage infiltration compared with wild-type mice after unilateral ureteral obstruction.26 The upregulation of Cfi and Rgs2 in the GBT1118-treated vs untreated sickle mice may highlight protective mechanisms against complement- and angiotensin-2–mediated damage in SCA and should be explored in future studies.
Hypoxia hallmark genes were predominantly downregulated in the glomeruli (Slc2a1 and Stc1) and kidney cortex (Csrp2, Bnip3l, Sdc3, Hexa, and Stc1) of the treated vs control sickle mice (P ≤ .01) (supplemental Table 4).
In conclusion, we demonstrate that reducing the degree of hemolysis with GBT1118, an analog of voxelotor, stabilizes kidney function in sickle mice. In patients with SCA, the presence of hemoglobinuria is associated with increased concentrations of urinary biomarkers of kidney injury (KIM-1), albuminuria, and progression of kidney disease.27,28 In this study, the GBT1118-treated sickle mice had improvements in markers of hemolysis that led to a reduction in urine Hb concentration and glomerular (nephrin) and tubular (KIM-1) injury biomarkers. This was followed by stabilization in the degree of hemosiderin deposition, inflammatory cell infiltration, interstitial fibrosis, and podocyte health, as well as preserved kidney function as assessed by urine albumin and protein concentration and by serum cystatin C in the GBT1118-treated vs control sickle mice. Limitations of our study include the use of TBARS, a nonspecific biomarker of lipid peroxidation, and the need to evaluate the effects of reduced hemolysis on vascular endothelial health, both of which we will plan to investigate in future studies. Subsequent studies should also examine the effects of GBT-1118 and improved hemolysis on preserving urine-concentrating ability and treating more advanced kidney disease in younger and older sickle mice, respectively. Developing strategies to mitigate hemolysis in patients with SCA has potential as a targeted treatment approach to improve sickle cell nephropathy.
Acknowledgments
The authors thank Global Blood Therapeutics for kindly providing the GBT1118 and control chow used in the experiments.
The project described was supported by the National Institutes of Health (NIH) through grant R01 HL-153161 (S.L.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: G.R., S.S., X.Z., A.S., M.A.R., and S.L.S. designed and performed research, analyzed the data, and wrote the paper; and R.D.M., J.P.L., and V.R.G. designed and performed research and wrote the paper.
Conflict-of-interest disclosure: V.R.G. and S.L.S. receive research support and provide consulting for Global Blood Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Santosh L. Saraf, Division of Hematology-Oncology, University of Illinois at Chicago, 820 South Wood St, Suite 172, Chicago, IL 60612; e-mail: ssaraf@uic.edu.

