

Key Points
Somatic UBA1 mutations define VEXAS in men with late-onset systemic inflammatory disease and cytopenia.
Features summarizing VEXAS include cytopenia, hypercellularity, lack of hematogones, and vacuoles in myeloid and erythroid precursors.
Abstract
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is caused by somatic mutations in UBA1 and is identified by a genotype-driven method. This condition affects unrelated men with adultonset inflammatory syndromes in association with hematologic manifestations of peripheral cytopenia and bone marrow myeloid dysplasia. Although bone marrow vacuolization restricted to myeloid and erythroid precursors has been identified in patients with VEXAS, the detailed clinical and histopathological features of peripheral blood and bone marrows remain unclear. The current case report describes the characteristic hematologic findings in patients with VEXAS, including macrocytic anemia, thrombocytopenia, marked hypercellular bone marrow with granulocytic hyperplasia, megaloblastic changes in erythroid precursors, and the absence of hematogones in addition to prominent vacuoles in myeloid and erythroid precursor cells. Characterizing the clinical and hematologic features helps to raise awareness and improve diagnosis of this novel, rare, but potentially underrecognized disease. Prompt diagnosis expands the general knowledgeable and understanding of this disease, and optimal management may prevent patients from developing complications related to this refractory inflammatory syndrome and improve the overall clinical outcome.
Introduction
Somatic mutations in hematopoietic stem progenitor cells (HSPCs) drive the pathogenesis of various hematologic diseases. Whole-genome sequencing and targeted deep sequencing have revealed a myriad of somatic mutations in myelodysplastic syndrome (MDS) and acute myeloid leukemia.1,2 Acquired mutations in HSPCs are also implicated in nonmalignant hematologic disorders. A classic example of the mutations is paroxysmal nocturnal hemoglobinuria, caused by clonal expansion of PIGA-mutated HSPCs.3 Clonal hematopoiesis (CH) is also frequently observed in acquired aplastic anemia.4 CH in healthy individuals without overt hematologic disease, also called clonal hematopoiesis of indeterminate potential, occurs with increasing age and is associated with an increased risk of coronary artery disease.5 More recently, somatic UBA1 mutations in HSPCs were reported in men presenting with severe adult-onset autoinflammatory disease, termed VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, and somatic syndrome).6 We report 2 patients presenting with systemic inflammatory disease, macrocytic anemia, and thrombocytopenia in the absence of cytogenetic or molecular abnormalities characteristic of a myeloid neoplasm. Genomic sequencing confirmed a sole UBA1 codon 41 mutation in both patients. We describe the clinical presentation and a detailed review of blood and bone marrow findings.
Case description
The first patient was a 63-year-old man presenting with recurrent fever, skin rash, and polyarthritis. Laboratory studies showed anemia, thrombocytopenia, markedly elevated C-reactive protein and erythrocyte sedimentation rate, and positive rheumatoid factor (Table 1). A skin biopsy specimen showed neutrophilic infiltration of papillary dermis consistent with Sweet syndrome (data not shown). Treatment with high-dose oral prednisone led to complete resolution of skin rash, arthritis, and other systemic symptoms. However, subsequent attempts to taper prednisone were unsuccessful, with prompt recurrence of symptoms despite treatment with several biologic agents (adalimumab, etanercept, abatacept, tocilizumab, anakinra, tofacitinib, and IV immunoglobin).
Summary of clinical presentation and pertinent laboratory experiments
| Overall clinical manifestations | Patient 1 | Patient 2 |
|---|---|---|
| Clinical presentation | ||
| Fever | Yes | Yes |
| Cytopenia | Yes | Yes |
| Arthritis | Inflammatory polyarthritis | Joint pain only |
| Dermatitis | Neutrophilic dermatitis (Sweet syndrome) | Leukocytoclastic vasculitis |
| Pneumonitis | No | Yes |
| CBC (normal range) | ||
| WBC, ×109/L (4.3-11.3) | 2.8 | 4.9 |
| Hemoglobin, g/dL (14.8-17.8) | 7.9 | 10.7 |
| MCV, fL (81.2-96.6L) | 110 | 114 |
| Platelets ×109/L (159-439) | 121 | 71 |
| ANC, ×109/L (2.0-7.4) | 2.5 | 2.9 |
| Laboratory (normal range) | ||
| C-reactive protein, mg/dL (0-0.8) | 11.2 | 10.7 |
| ESR, mm/h (0-10) | 105 | 62 |
| Bone marrow | ||
| Circulating blasts (%) | 0 | 0 |
| Cellularity (%) | 95 | 95 |
| Blasts (%) | 1 | 4 |
| Myeloid/erythroid ratio | 2.5 | 5.6 |
| Ring sideroblasts (%) | 5 | 0 |
| Reticulin stain | MF 1/3 | MF 1/3 |
| Ancillary studies | ||
| Karyotype | Normal | Normal |
| Variants by myeloid NGS panel (VAF %) | PRPF40B p.E399D (48.4) | None |
| UBA1 variants (VAF %) | p.M41L (67.3) | p.M41V (83.6) |
| Overall clinical manifestations | Patient 1 | Patient 2 |
|---|---|---|
| Clinical presentation | ||
| Fever | Yes | Yes |
| Cytopenia | Yes | Yes |
| Arthritis | Inflammatory polyarthritis | Joint pain only |
| Dermatitis | Neutrophilic dermatitis (Sweet syndrome) | Leukocytoclastic vasculitis |
| Pneumonitis | No | Yes |
| CBC (normal range) | ||
| WBC, ×109/L (4.3-11.3) | 2.8 | 4.9 |
| Hemoglobin, g/dL (14.8-17.8) | 7.9 | 10.7 |
| MCV, fL (81.2-96.6L) | 110 | 114 |
| Platelets ×109/L (159-439) | 121 | 71 |
| ANC, ×109/L (2.0-7.4) | 2.5 | 2.9 |
| Laboratory (normal range) | ||
| C-reactive protein, mg/dL (0-0.8) | 11.2 | 10.7 |
| ESR, mm/h (0-10) | 105 | 62 |
| Bone marrow | ||
| Circulating blasts (%) | 0 | 0 |
| Cellularity (%) | 95 | 95 |
| Blasts (%) | 1 | 4 |
| Myeloid/erythroid ratio | 2.5 | 5.6 |
| Ring sideroblasts (%) | 5 | 0 |
| Reticulin stain | MF 1/3 | MF 1/3 |
| Ancillary studies | ||
| Karyotype | Normal | Normal |
| Variants by myeloid NGS panel (VAF %) | PRPF40B p.E399D (48.4) | None |
| UBA1 variants (VAF %) | p.M41L (67.3) | p.M41V (83.6) |
Semiquantitative grading of reticulin fibrosis was performed using the MF grading system as described in the current WHO classification.8
ANC, absolute neutrophil count; CBC, complete blood count; ESR, erythrocyte sedimentation rate; MCV, mean corpuscular volume; MF marrow fibrosis; WBC, white blood cell.
The second patient was a 67-year-old man presenting with fever, body aches, and progressive dyspnea with minimal exertion. Laboratory findings were similar to patient 1 (Table 1). Chest computed tomography demonstrated diffuse bilateral ground glass opacities and small pleural effusions. Systemic symptoms improved with oral prednisone and, similar to patient 1, taper of prednisone resulted in recurrence of symptoms. He subsequently developed palpable purpura and symptomatic extensive left leg great saphenous vein thrombus. A skin biopsy specimen showed superficial perivascular and mild inflammatory infiltrate with extravasated erythrocytes, suggestive of leukocytoclastic vasculitis (data not shown). The patient continued to be treated with oral prednisone, 10 mg twice daily, and was receiving continuous oxygen supplementation at last follow-up.
Methods
Wright‐Giemsa, iron, hematoxylin and eosin, and reticulin staining and immunohistochemistry (antibodies against CD34, QBEnd/10; Leica Biosystems Inc, Lincolnshire, IL; and myeloperoxidase; Polyclonal, Santa Clara, CA) were performed on blood smear slides, bone marrow aspirate, and core biopsy sections according to standard protocols. All slides were examined by P.L., K.M., T.G., and R.R.M. independently. Flow cytometric, cytogenetic analyses, and a myeloid malignancy panel by next-generation sequencing (NGS) were performed as part of the standard protocol for myeloid neoplasm workup at ARUP Laboratories. Targeted hybrid‐capture sequencing was performed on 64 genes (supplementary Table 1), by using the standard protocol established at ARUP for enrichment, library preparation, sequencing, and data analysis.7 Patients consented to an National Institutes of Health research protocol (NCT00001373) and DNA obtained from peripheral blood mononuclear cells was subjected to Sanger sequencing and droplet digital polymerase chain reaction assay for UBA1 mutations.6
Results and discussion
Blood and bone marrow morphologic features were identical in both patients. Blood film examination showed macrocytic red cells with normal appearing leukocytes (Figure 1A-B). Hypercellular bone marrow was seen (Figure 1C-D) with an absence of immature B cells (hematogones, data not shown) suggesting a chronic bone marrow injury. Marked granulocytic hyperplasia was seen on morphology and immunohistochemistry (Figure 1D-E). The blast count was normal by morphology or CD34 immunostain (Figure 1F) with no significant reticulin fibrosis (data not shown). The erythroid precursors showed marked vacuolization with megaloblastic changes (Figure 1G-H) without nuclear membrane abnormalities, and rare ring sideroblasts (<5% of total erythroid cells) were seen in patient 1 (data not shown). Striking vacuolization was also evident in myeloblasts, promyelocytes, and immature monocytes (Figure 1I-K). Occasional immature megakaryocytes with increased nuclear/cytoplasmic ratios were observed (Figure 1L). No overt dysplasia was observed, and morphologic findings did not meet the World Health Organization MDS criteria.8 Flow cytometric analyses demonstrated an absence of hematogones, polytypic B, and plasma cells, and no aberrant T or NK cells (data not shown). Interestingly, aberrant maturation of neutrophils was observed in patient 2 (Figure 1M) with normal monocytes and myeloblasts (data not shown). Overall, morphologic and immunophenotypic features were nonspecific and were seen in myeloid neoplasms or as reactive changes in response to systemic inflammation, among others. Karyotyping revealed normal male chromosomal complements in both patients. Multiple targeted NGS showed a single variant of uncertain significance in the PRPF40B gene in patient 1 at a persistent near-heterozygous variant allele frequency (VAF) of 48%, suggesting likely germline origin and no variants present in patient 2. Sanger sequencing confirmed a UBA1 p.Met41Leu mutation in patient 1 and UBA1 p.Met41Val mutation in patient 2 (Figure 1N). Droplet digital polymerase chain reaction assay revealed a VAF of 67.3% and 83.6% in patients 1 and 2, respectively (Table 1).
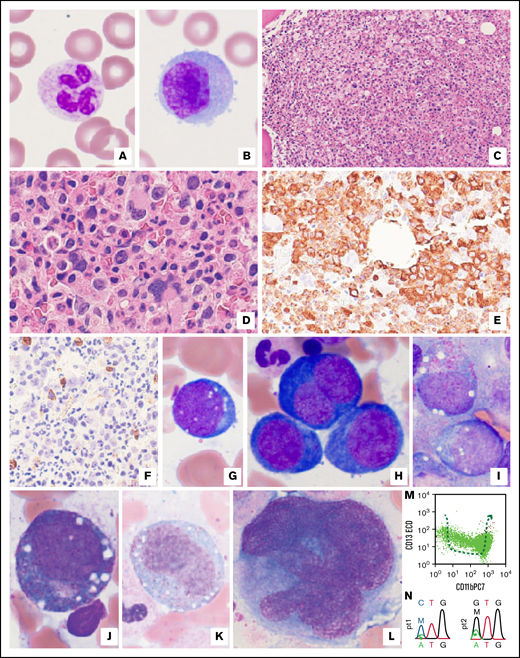
Examination of peripheral blood and bone marrow biopsy specimens, as well as ancillary studies for both VEXAS patients. (A-B) There were no cytoplasmic vacuoles or overt dysplastic changes in neutrophils (A) or monocytes (B) in peripheral blood (representative pictures from patient 1) Original magnification ×1000. (C-E) Markedly hypercellular bone marrow (C) showed extensive granulocytic hyperplasia with complete maturation (D). (E) This effect is also illustrated by MPO immunostaining in a representative image from patient 2. Original magnifications ×200 (C), ×400 (D-E). (F) By immunohistochemistry, there was no significant increase in CD34+ blasts (3% to 4% of bone marrow elements). Original magnification ×400, a representative image from patient 2. (G-H) Characteristic vacuoles were found in erythroid precursors (pronormoblasts) (G), and profound megaloblastic changes were seen (H) in patient 1, although overt nuclear membrane irregularity was not appreciated. Original magnification ×1000. (I-K) Prominent vacuoles were also identified in myeloblasts (I), promyelocytes (J), and immature monocytes (K) in patient 2. Original magnification ×1000. (L) Frequent immature-appearing megakaryocytes showed an increased nuclear/cytoplasmic ratio, whereas typical dysplastic changes were not identified in patient 2. Original magnification ×1000. (M) By flow cytometry, granulocytes exhibited an abnormal maturation pattern illustrated by plots with CD13 vs CD11b in patient 2. (M) Dashed arrow indicates the normal granulocytic maturation pathways. (N) UBA1 mutations p.M41L and p.M41V were confirmed by Sanger sequencing in patients 1 and 2, respectively. Top codons, patients’ variants; bottom codons, reference codons. M, mutant; W, wild-type.
Examination of peripheral blood and bone marrow biopsy specimens, as well as ancillary studies for both VEXAS patients. (A-B) There were no cytoplasmic vacuoles or overt dysplastic changes in neutrophils (A) or monocytes (B) in peripheral blood (representative pictures from patient 1) Original magnification ×1000. (C-E) Markedly hypercellular bone marrow (C) showed extensive granulocytic hyperplasia with complete maturation (D). (E) This effect is also illustrated by MPO immunostaining in a representative image from patient 2. Original magnifications ×200 (C), ×400 (D-E). (F) By immunohistochemistry, there was no significant increase in CD34+ blasts (3% to 4% of bone marrow elements). Original magnification ×400, a representative image from patient 2. (G-H) Characteristic vacuoles were found in erythroid precursors (pronormoblasts) (G), and profound megaloblastic changes were seen (H) in patient 1, although overt nuclear membrane irregularity was not appreciated. Original magnification ×1000. (I-K) Prominent vacuoles were also identified in myeloblasts (I), promyelocytes (J), and immature monocytes (K) in patient 2. Original magnification ×1000. (L) Frequent immature-appearing megakaryocytes showed an increased nuclear/cytoplasmic ratio, whereas typical dysplastic changes were not identified in patient 2. Original magnification ×1000. (M) By flow cytometry, granulocytes exhibited an abnormal maturation pattern illustrated by plots with CD13 vs CD11b in patient 2. (M) Dashed arrow indicates the normal granulocytic maturation pathways. (N) UBA1 mutations p.M41L and p.M41V were confirmed by Sanger sequencing in patients 1 and 2, respectively. Top codons, patients’ variants; bottom codons, reference codons. M, mutant; W, wild-type.
Similar to the VEXAS cohort reported originally, examination of the bone marrow of our patients showed vacuolization of myeloid and erythroid progenitors without features characteristic of a myeloid neoplasm. Other common causes of bone marrow vacuolation were considered including copper deficiency, toxin-induced bone marrow changes, and genetic syndromes such as Pearson’s syndrome and Menkes disease (supplementary Table 2).9-12 Bone marrow changes in copper deficiency are distinguishable based on a hypocellular bone marrow, granulocytic hypoplasia, relative erythroid hyperplasia, and preserved megakaryopoiesis.12 Moreover, increased stainable iron and mild dyserythropoiesis with occasional ring sideroblasts may also be seen in copper deficiency. Pearson’s syndrome is characterized by vacuolated myeloid and erythroid progenitors and increased ring sideroblasts, and an early childhood presentation with diarrhea and cytopenias is typical.11 Vacuoles in myeloid and erythroid precursors can also be seen in acute alcohol ingestion and drug-induced bone marrow damage (supplementary Table 2).13,14 Vacuoles in hematopoietic precursors are extremely rare in MDS.15 The clinical presentation of our patients was similar to that of the National Institutes of Health cohort, with male sex, adult onset, and dependence on corticosteroids without clinical response to other biologic agents.6
VEXAS is a unique systemic autoinflammatory disease caused by somatic UBA1 mutations in the HSPCs that affect men exclusively. UBA1 is an X-linked gene necessary for the initiation of ubiquitylation, a posttranslational protein modification process that involves the conjugation of ubiquitin to the substrate protein's lysine residues. Ubiquitin-activating (E1), conjugating (E2), and ligating (E3) enzymes act sequentially during ubiquitylation.16,17 The ubiquitin system regulates innate immune-signaling pathways and plays a crucial role in adaptive immune responses and immune tolerance.18 Thus, dysregulated ubiquitylation can lead to a systemic inflammatory response. T lymphocytes have an indispensable role in immune tolerance, and somatic mutations in T cells have long been recognized in association with autoimmune disorders such as rheumatoid arthritis.19 The association of systemic inflammation with somatic mutation restricted to peripheral blood granulocytes and monocytes with wild-type T and B cells is intriguing. In the future, classification of certain autoimmune diseases, such as relapsing polychondritis, may include the presence of UBA1 mutations, as these patients often are dependent on corticosteroids.20 Interestingly, ∼33% of patients with VEXAS were diagnosed with MDS by disease-defining cytogenetic or molecular aberrancies.21 Comparing the incidence to that in age-matched general population, somatic UBA1 mutation predisposes these patients to MDS and other associated hematologic neoplasms, requiring close clinical follow-up.21 Potential future therapies could include targeting the ubiquitylation pathways, UBA1 gene editing to alter the mutant clone, or allogeneic bone marrow transplantation to replace affected progenitor cells.22,23
Authorship
Contribution: P.L., S.V., and S.K.T. designed the study and drafted the manuscript; T.K. and S.K.T. examined patients and provided patients’ clinical history; P.L., K.A.M., R.R.M., and T.G. performed the bone marrow examination; D.O.C., L.W., and D.B.B. performed the molecular experiments for UBA1 variants; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peng Li, Department of Pathology, 15 North Medical Dr, University of Utah, Salt Lake City, UT 84108; e-mail, pengl.li@aruplab.com.

