

Key Points
MDM2 inhibitors have potent in vivo activity against and could be a novel therapy for EBV-positive B-cell lymphoma.
EBV positivity or loss of BCL6 expression can be a potential predictive biomarker for response to MDM2 inhibitors in patients with lymphoma

Abstract
Epstein-Barr virus–positive (EBV-positive) B-cell lymphomas are common in immunocompromised patients and remain an unmet medical need. Here we report that MDM2 inhibitors (MDM2is) navtemadlin and idasanutlin have potent in vivo activity in EBV-positive B-cell lymphoma established in immunocompromised mice. Tumor regression was observed in all 5 EBV-positive xenograft–associated B-cell lymphomas treated with navtemadlin or idasanutlin. Molecular characterization showed that treatment with MDM2is resulted in activation of p53 pathways and downregulation of cell cycle effectors in human lymphoma cell lines that were either EBV-positive or had undetectable expression of BCL6, a transcriptional inhibitor of the TP53 gene. Moreover, treatment with navtemadlin resulted in tumor regression and prevented systemic dissemination of EBV-positive lymphoma derived from 2 juvenile patients with posttransplant lymphoproliferative diseases, including 1 whose tumor was resistant to virus-specific T-cell therapy. These results provide proof-of-concept for targeted therapy of EBV-positive lymphoma with MDM2is and the feasibility of using EBV infection or loss of BCL6 expression to identify responders to MDM2is.
Introduction
Epstein-Barr virus (EBV), a human gamma-herpesvirus, is etiologically associated with a variety of lymphoproliferative diseases and malignant lymphomas.1 EBV-positive B-cell lymphomas are particularly common in immunocompromised individuals, including those with posttransplant lymphoproliferative disease (PTLD), HIV-associated lymphoma, lymphoma of the elderly, and lymphoma in patients with primary immunodeficiency syndromes or those being treated with immunosuppressive therapies.2 The major mechanisms underlying the outgrowth of PTLD are loss of EBV-specific T-cell immunity and uninhibited growth of EBV-infected lymphocytes in an immunocompromised environment.3,4 Treatment options for most patients with EBV-positive lymphoma are the same as those for lymphoma in immunocompetent patients except that reduction in immunosuppression may be used for treating patients who are receiving immunosuppressive therapy. For refractory EBV-positive lymphoma, adoptive immunotherapy with EBV-specific T cells has been tested in clinical trials over the past 25 years, and several studies showed promising results.5,6 However, this therapy is only available at hospitals performing such clinical trials, and resistance to adoptive T-cell therapy has been observed.7 Overall, most patients with EBV-positive B-cell lymphoma are individuals with a certain degree of immunodeficiency who have worse survival than immunocompetent patients with lymphoma.8,9 Estimated 5-year overall survival (OS) rates were 25% in patients with EBV-positive diffuse large B-cell lymphoma (DLBCL) but were 65% in EBV-negative patients.9,10
With overall incidence rates of PTLD ranging from 1% to 20% in solid organ transplant recipients3 and at about 3.5% in hemopoietic stem cell transplant recipients,11 PTLD is one of the common life-threatening complications of solid organ transplants and allogeneic hemopoietic stem cell transplants.3 Most PTLDs are monomorphic, with DLBCL being the most frequent subtype, and about 90% of patients with PTLD are EBV positive.2 Current treatment of PTLD includes reduction in immunosuppression, rituximab (anti-CD20 antibody), and conventional chemotherapy.3 With all currently available treatment options, the median OS duration in solid organ transplant patients with PTLD is about 6.6 years.8 Similar to lymphoma in patients with PTLD, lymphoma is the leading cause of cancer morbidity and mortality in HIV-infected patients who have increased incidence of EBV-positive lymphoma despite the use of optimal combined antiretroviral therapy.12 For all treatment options available, the 5-year OS rates for DLBCL in HIV-infected and uninfected patients were about 41% and 61%, respectively.13 Altogether, for the majority of patients with EBV-positive PTLD and lymphoma, significant unmet medical needs remain. Therefore, development of new therapeutic strategies or agents for treatment in these subgroups of patients is urgently needed. Here we report that E3 ubiquitin protein ligase MDM2 inhibitors (MDM2is) navtemadlin (formerly AMG-232 or KRT-232) and idasanutlin have potent in vivo activity in EBV-positive B-cell lymphoma, triggering tumor regression and preventing systemic dissemination of lymphomas established in immunocompromised mice. Our results suggested the feasibility of using EBV infection or loss of BCL6 expression as a potential predictive biomarker for response to MDM2is in patients with lymphoma.
Methods
Therapeutic agents
Navtemadlin was provided by Amgen and Kartos Therapeutics through the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. Idasanutlin and trametinib were obtained from Selleck Chemicals.
Xenograft-associated B-cell lymphoma (XABCL), lymphoma cell lines, and PTLD cells from patients
Established cell lines were obtained from American Type Culture Collection and maintained in our laboratories. TRL1 and TRL595 were cultured from tumors of juvenile patients with PTLD after bone marrow transplant7 and liver transplant,14 respectively, as previously reported. XABCL cells were obtained during our study to establish patient-derived xenografts (PDXs) from clinical specimens from patients with non-small cell lung cancer (NSCLC). The origin of XABCL was verified by DNA fingerprint together with the patients’ genomic DNA from peripheral blood mononuclear cells or tumor samples, as previously described.15,16 All clinical samples and data were collected with informed consent of the patients under a research protocol approved by the Institutional Review Board at The University of Texas MD Anderson Cancer Center (MDACC). All PDXs were established from fresh lung cancer samples that were collected from surgically resected specimens under research protocols approved by the Institutional Review Board at MDACC. All clinical samples and data were collected with informed consent from the patients.
Next-generation sequencing
Whole-exome sequencing and RNA sequencing (RNA-seq) analyses were performed at the Advanced Technology Genomics Core at MD Anderson. Genomic libraries were prepared and whole-exome regions were captured using the SureSelect Low Input Target Enrichment System Clinical Research Exome version 2 (Agilent Technologies), following the manufacturer’s protocol. The captured libraries were sequenced on a HiSeq 4000 (Illumina) on a version 3 TruSeq paired-end flow cell, according to the manufacturer’s instructions. For RNA-seq analysis, RNA was isolated from samples, and libraries were prepared from RNA samples using the NuGEN Ovation RNA-seq system, following the manufacturer’s instructions. The libraries were then sequenced on the NovaSeq 6000 system (Illumina) using paired-end run format. RNA-seq data processing was performed at the Cancer Genomics Laboratory at our institution. Detailed methods for data analyses are provided in the supplemental Methods.
RPPA and cell viability assays
The reverse phase protein array (RPPA) assay was performed at the Functional Proteomics RPPA Core facility at MDACC, as previously reported.17,18 Detailed methods are provided in the supplemental Methods. Two-way analysis of variance (ANOVA) was used to find genes that were expressed differentially between treatment and control samples. We used the Benjamini-Hochberg procedure to control the false discovery rate (FDR) in multiple testing experiments.19 Differentially expressed genes were defined as P ≤ .05 at FDR ≤ 0.05. Heatmaps were made using only the differentially expressed genes. Cell viability was determined by using 3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium inner salt (MTS) and the electron coupling reagent phenazine ethosulfate (PES), as previously described.20
Immunohistochemical staining and western blot analysis
Antibodies used in this study are listed in supplemental Table 1. Histologic characterization of XABCL sections was performed by hematoxylin and eosin staining and immunohistochemical staining with antibodies against human CD20, as previously reported.15 Western blot analysis was performed as previously described.20
Animal experiments
Each tumor was subcutaneously inoculated into the dorsal flanks of female nude mice or NSG mice (non-obese diabetic/severe combined immunodeficiency [NOD-SCID] mice with null mutations of the gene encoding interleukin-2 receptor γ). Subcutaneous tumors were measured with calipers, and tumor volume was calculated according to the formula V = ab2/2, where a is the largest diameter and b is the smallest. Mice were randomly assigned to treatment groups (n = 3-5 mice per group) when tumors reached 200 mm3 (about 7-9 mm in diameter). Records of treatment and results were not kept in a blinded manner. Tumor volume changes were calculated with the R software program, with tumor volume at the beginning of treatment set to a baseline of 0, as previously reported.21 When the tumors reached 15 mm in diameter, the experiment was ended, and the mice were euthanized. All animal studies were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-23) and the institutional guidelines of the MDACC.
Statistical analysis
Statistical analysis for difference among groups was performed with ANOVA or 2-sample Student t tests, as reported previously.21 Mutations in XABCLs and matched NSCLC tumor samples were compared by using Maftools.22 For in vivo studies, changes in tumor volume and adjusted area under the curve at given time points were compared between the treatment and control groups, if applicable. A multiplicity adjustment was performed by controlling the FDR, as described previously.19 All statistical analyses were performed using R software version 3.6.2. A P value of < .05 was regarded as significant.
Results
MDM2is trigger regression of EBV-positive XABCL in vivo
We and others previously reported that proliferation of EBV-infected B cells infiltrated in human tumor specimens that were implanted in immunocompromised mice led to the formation of XABCL.15,23 The XABCL tumors predominantly consisted of human CD19+ and CD20+ B cells, with histologic features of DLBCL and high copy numbers of the EBV-encoded gene EBNA1 when analyzed by polymerase chain reaction. To elucidate the evolution of primary tumors into XABCLs, we analyzed genomic alterations of 25 XABCLs by whole-exome sequencing and compared the results with the matched NSCLC tumors from which the XABCLs were derived. TP53 mutation was detected in 9 (36%) of 25 primary NSCLC tumors but only 2 (8%) of 25 of XABCLs (Figure 1A; supplemental Table 2). This result supported the previous reports that EBV-positive posttransplant DLBCL has a low mutation frequency in the TP53 gene.24,25 In contrast, mutations in FSIP2, ZNF107, DICER1, IGLL5, KMT2D, and ARID1A were more frequent in XABCLs than in their corresponding primary tumors. The significance of mutations in these genes in XABCLs is not clear. Nevertheless, mutations of the KMT2D and ARID1A genes are relatively common in EBV-positive Burkitt lymphoma.26 We also determined the expression of oncogenic viral genes in RNA-seq data available from 15 XABCL samples. Of 7 oncogenic viruses searched for their RNA transcripts, only type 1 and type 2 EBV-specific RNA reads were detected in XABCL samples. All 15 XABCL samples expressed LMP1 from type 1 and/or type 2 EBVs, albeit at variable levels (Figure 1B; supplemental Table 3). EBNA2 was expressed in all 15 XABCLs, 14 from type 1 EBV and 1 from type 2 EBV. This result indicates that most EBV-positive XABCLs had type II or type III EBV latency. Type II and type III EBV latencies are also the most common types of EBV latency observed in patients with PTLD and EBV-positive DLBCL.9,27 These results indicated that XABCLs recapitulated certain molecular and pathologic features of EBV-positive lymphoma.
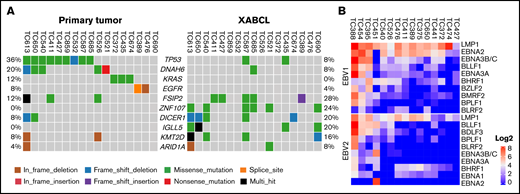
Molecular characterization of XABCLs. (A) Gene mutations in 25 XABCLs and the matched NSCLC tumors from which the XABCLs were derived. Only samples with mutations shown in the co-oncoplot were presented in the graph. Detailed mutation information for 25 pairs of samples is shown in supplemental Table 2. (B) RNA transcripts of type I and type II EBV detected in RNA-seq data of 15 XABCLs. LMP1 and EBNA2 were detected in all 15 samples, suggesting that XABCLs have type II or type III latency of EBV infection.
Molecular characterization of XABCLs. (A) Gene mutations in 25 XABCLs and the matched NSCLC tumors from which the XABCLs were derived. Only samples with mutations shown in the co-oncoplot were presented in the graph. Detailed mutation information for 25 pairs of samples is shown in supplemental Table 2. (B) RNA transcripts of type I and type II EBV detected in RNA-seq data of 15 XABCLs. LMP1 and EBNA2 were detected in all 15 samples, suggesting that XABCLs have type II or type III latency of EBV infection.
In an effort to develop effective therapies for TP53 wild-type (wt) lung cancers, we recently evaluated effects of combination therapy of MDM2i navtemadlin and MEK inhibitor trametinib in 22 NSCLC PDX models.21 One of the XABCLs derived from NSCLC (TC388) was also included in that study. Both NSCLC PDXs and XABCLs were established in immunocompromised mice. Interestingly, TC388, which has wt TP53 with DLBCL histology and a high EBV DNA copy number, showed an extraordinary response to oral gavage administration of both single-agent therapy with navtemadlin and combination therapy of navtemadlin plus trametinib, both resulting in tumor regression >90% (Figure 2). We then tested the activity of navtemadlin in 4 other XABCL models. The results showed all XABCLs exhibited similar dramatic responses to single-agent therapy with navtemadlin. Most mice had complete or >50% tumor regression by the end of 3 weeks of treatment and, for most mice, no tumor regrowth was observed for 30 days after treatment stopped. In 1 of the models (TC859) treated with navtemadlin, tumors regrew at 70 to 80 study days but responded to a second cycle of treatment with navtemadlin. In another of the XABCL models (TC389), we tested single-agent activity of navtemadlin and idasanutlin (another MDM2i28 ) and found similar anticancer activity between the 2 agents (Figure 2). These results demonstrated that single-agent therapy with navtemadlin or idasanutlin was sufficient to induce tumor regression in XABCL models.
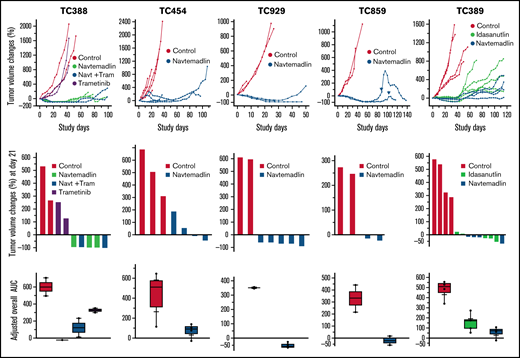
Tumor volume changes of 5 XABCLs after treatment with MDM2is. Mice were treated with vehicle, 45 mg/kg of navtemadlin (Navt), or 25 mg/kg idasanutlin by oral gavage once a day for 5 days per week for 3 weeks or with 0.1 mg/kg of trametinib (Tram) by oral gavage once a day for 4 weeks. Top: tumor volume changes in individual mice bearing 1 of 5 XABCLs as indicated. Middle: waterfall graphs showing tumor volume changes at the end of treatment (day 21). Bottom: adjusted area under curve (AUC) for entire study time period. The values represent the median (line inside box) and the third and first quartile (box) ±1.5× the interquartile range from the top and bottom of the box (error bar). P < .05 for all treatment groups except trametinib when compared with the control. Of note, arrows on the growth curve of TC859 indicate the second cycle of treatment after tumor regrew at 70 to 80 study days. For model TC389, treatment with idasanutlin was also tested. Results showed that both navtemadlin and idasanutlin can significantly inhibit XABCL in vivo.
Tumor volume changes of 5 XABCLs after treatment with MDM2is. Mice were treated with vehicle, 45 mg/kg of navtemadlin (Navt), or 25 mg/kg idasanutlin by oral gavage once a day for 5 days per week for 3 weeks or with 0.1 mg/kg of trametinib (Tram) by oral gavage once a day for 4 weeks. Top: tumor volume changes in individual mice bearing 1 of 5 XABCLs as indicated. Middle: waterfall graphs showing tumor volume changes at the end of treatment (day 21). Bottom: adjusted area under curve (AUC) for entire study time period. The values represent the median (line inside box) and the third and first quartile (box) ±1.5× the interquartile range from the top and bottom of the box (error bar). P < .05 for all treatment groups except trametinib when compared with the control. Of note, arrows on the growth curve of TC859 indicate the second cycle of treatment after tumor regrew at 70 to 80 study days. For model TC389, treatment with idasanutlin was also tested. Results showed that both navtemadlin and idasanutlin can significantly inhibit XABCL in vivo.
MDM2is inhibited cell cycle effectors that were upregulated upon EBV-mediated reprogramming of B lymphocytes
We characterized molecular changes induced by MDM2i treatment in vivo. For this purpose, NSG mice (n = 3 mice per group) bearing XABCLs TC389 and TC929 were treated with vehicle, navtemadlin, or idasanutlin for 3 days. Proteomic changes in tumor samples were determined by RPPA assay for 483 proteins or protein phosphorylation sites. Protein levels of p21, SDHA, and p53 were dramatically increased in both models after treatment with either navtemadlin or idasanutlin compared with solvent control. In contrast, protein levels of CDC25C, aurora kinase B (AURKB), RRM2, FOXM1, PLK1, cyclin B, CHEK1, and phosphorylated RB-pS807/811 and P90RSK-pT573 were significantly downregulated (adjusted P < .05; Figure 3A; supplemental Table 4). Most of these downregulated proteins promote cell cycle progression and have been investigated as anticancer targets. Interestingly, AURKB,29 PLK1,30 CHEK1,31 and CDC25C32 have been previously reported to be upregulated in EBV-transformed B lymphocytes or to play a role in EBV-mediated B-cell lymphomagenesis. We therefore analyzed expression changes of genes whose proteins were altered in the RPPA analysis in longitudinal RNA-seq data for EBV-mediated transcriptional reprogramming in B lymphocytes reported previously by Merozke-Gorska et al.33 Cyclin B1 (CCNB1), RRM2, AURKB, FOXM1, PLK1, CHEK1, and CDC25C were all significantly upregulated over time in B lymphocytes after EBV infection (Figure 3B). These results demonstrated that a number of cell cycle effectors that were upregulated by EBV infection in B lymphocytes were suppressed by treatment with MDM2is.
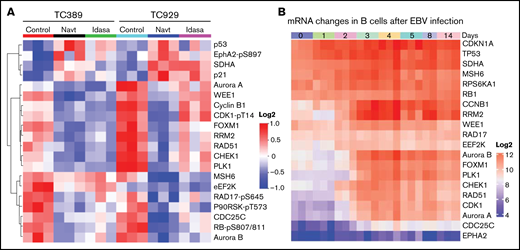
Molecular changes induced in XABCL in vivo. (A) Mice bearing XABCLs TC389 and TC929 (n = 3 mice per group) were treated with solvent, navtemadlin, or idasanutlin (Idasa) for 3 days. Tumors were harvested for RPPA analysis as described in “Methods.” Proteins with significant changes (adjusted P < .05) induced by both MDM2is in both models are shown in the heatmap. (B) Change in mRNA induced during B-lymphocyte reprogramming after EBV infection. The data were downloaded from http://ebv-b.helmholtz-muenchen.de/. Heatmap shows genes whose proteins were included in panel A.
Molecular changes induced in XABCL in vivo. (A) Mice bearing XABCLs TC389 and TC929 (n = 3 mice per group) were treated with solvent, navtemadlin, or idasanutlin (Idasa) for 3 days. Tumors were harvested for RPPA analysis as described in “Methods.” Proteins with significant changes (adjusted P < .05) induced by both MDM2is in both models are shown in the heatmap. (B) Change in mRNA induced during B-lymphocyte reprogramming after EBV infection. The data were downloaded from http://ebv-b.helmholtz-muenchen.de/. Heatmap shows genes whose proteins were included in panel A.
Activity of MDM2is in human B-cell lymphoma cell lines in vitro
To test whether human EBV-positive lymphoma cells are sensitive to MDM2is, we first tested navtemadlin on cells from the Burkitt lymphoma cell line Raji compared with 2 cell lines established from the XABCLs TC389 and TC526. TC389 and TC526 cells were moderately sensitive to navtemadlin in vitro, with a half maximal inhibitory concentration (IC50) of about 1 µM, whereas the Raji cells were resistant to navtemadlin with IC50 >10 µM. Because Raji cells are TP53 mutant, their resistance to navtemadlin is not unexpected. We then determined the sensitivity of 2 TP53-wt mantle cell lymphoma lines, JVM2 and Z138. JVM2 is EBV positive, whereas the EBV status of Z138 is unknown. The viability analysis showed that these 2 cell lines have similar or greater sensitivity to navtemadlin and idasanutlin compared with TC526 and TC389 cells (Figure 4A-B). We also determined expression of p53, p21, and EBV-encoded LMP1 in these cell lines. A treatment-associated increase in p53 was detected in cell lysates of TC526 and JVM2 but not in Raji and Z138 cells harvested at 24 hours after treatment with navtemadlin or idasanutlin (Figure 4C). EBV viral protein LMP1 was detected in TC526, Raji, and JVM2 cells, confirming their positivity for EBV, but not in Z138 cells.
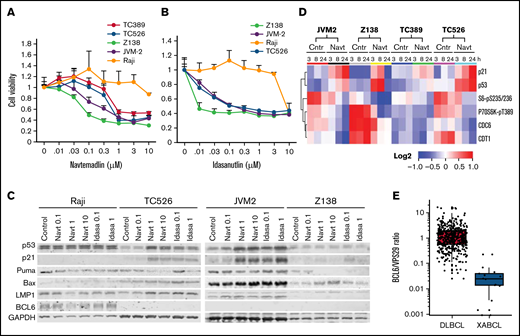
Effects of MDM2is in human lymphoma cell lines in vitro. (A-B) Dose response to MDM2is. Cells were treated with (A) navtemadlin or (B) idasanutlin at the indicated doses for 3 days, and then cell viability was measured by assay with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). Cells treated with dimethyl sulfoxide were the control (Cntr), and their viability was set as 1. Each experiment was performed in quadruplicate and repeated at least twice. Values are means for quadruplicate assays; error bars are standard deviations (SDs). (C) Western blot analysis for proteins at the basal level and cells harvested 24 hours after treatment. (D) Heatmap of treatment-induced protein changes. Cells derived from XABCLs and human lymphoma cell lines were treated with solvent or 1 µM navtemadlin for 3, 8, or 24 hours, and cell lysates were subjected to RPPA analysis. The levels of proteins that had significant difference after treatment with solvent control and navtemadlin (adjusted P < .05) are presented in the heatmap scaled from low to high as blue-white-red. (E) BCL6 levels in RNA-seq data of 15 XABCLs and 480 patients with DCLBL from the NCICCR in The Cancer Genome Atlas database. Levels of BCL6 in each sample were normalized with total numbers of reads mapped to the human genome and then normalized with housekeeping gene VPS29 as indicated. The values represent the median (line inside box) and the third and first quartile (box) ±1.5 × the interquartile range from the top and bottom of the box (error bar), whereas the dots represent individual values.
Effects of MDM2is in human lymphoma cell lines in vitro. (A-B) Dose response to MDM2is. Cells were treated with (A) navtemadlin or (B) idasanutlin at the indicated doses for 3 days, and then cell viability was measured by assay with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). Cells treated with dimethyl sulfoxide were the control (Cntr), and their viability was set as 1. Each experiment was performed in quadruplicate and repeated at least twice. Values are means for quadruplicate assays; error bars are standard deviations (SDs). (C) Western blot analysis for proteins at the basal level and cells harvested 24 hours after treatment. (D) Heatmap of treatment-induced protein changes. Cells derived from XABCLs and human lymphoma cell lines were treated with solvent or 1 µM navtemadlin for 3, 8, or 24 hours, and cell lysates were subjected to RPPA analysis. The levels of proteins that had significant difference after treatment with solvent control and navtemadlin (adjusted P < .05) are presented in the heatmap scaled from low to high as blue-white-red. (E) BCL6 levels in RNA-seq data of 15 XABCLs and 480 patients with DCLBL from the NCICCR in The Cancer Genome Atlas database. Levels of BCL6 in each sample were normalized with total numbers of reads mapped to the human genome and then normalized with housekeeping gene VPS29 as indicated. The values represent the median (line inside box) and the third and first quartile (box) ±1.5 × the interquartile range from the top and bottom of the box (error bar), whereas the dots represent individual values.
To detect early molecular changes induced by navtemadlin in XABCL cells and in JVM2 and Z138 in vitro, we treated these cells with dimethyl sulfoxide solvent or 1 µM navtemadlin and harvested them at 3, 8, and 24 hours after treatment. Cell lysates were submitted to RPPA analysis with 483 validated antibodies similar to our procedure for in vivo tumor samples (supplemental Table 5). Two-way ANOVA was used to determine differences in protein levels associated with treatments among the cell lines. Figure 4D showed proteins significantly altered in all 4 cell lines (adjusted P < .05). Notably, p53 and p21 were significantly upregulated in all 4 cell lines at 3 hours after the treatment. In contrast, CDT1, CDC6, phospho-S6, and phospho-P70S6k were dramatically downregulated, suggesting that DNA replication initiation, protein synthesis, and G1/S cell cycle transition were the early events affected by treatment with navtemadlin. Of note, p53 and p21 were significantly upregulated in Z138 cells at 3 and 8 hours after treatment with navtemadlin, but not at 24 hours (Figure 4D), consistent with western blot analysis, in which upregulation of p53 or p21 in Z138 cells was barely detectable at 24 hours after treatment with MDM2is (Figure 4C). Altogether, these results indicated that treatment with navtemadlin induced similar molecular changes in the sensitive human B-cell lymphoma cell lines and XABCL-derived cell lines.
Although MDM2 is a major inactivator of the p53 protein,34,35 p53 is also inhibited by BCL636,37 transcriptionally. Interestingly, BCL6 expression is known to be strongly inhibited in EBV-infected DLBCL.38-40 To better understand EBV-mediated alteration of BCL6 expression, we examined BCL6 messenger RNA (mRNA) levels in publicly available RNA-seq data from a longitudinal study of newly infected naïve human B lymphocytes.33 BCL6 mRNA was significantly downregulated at all time points after the second day of EBV infection in human B lymphocytes (P < .0001; supplemental Figure 1). We then compared BCL6 levels in RNA-seq data between 15 XABCLs and 482 patients with DLBCL (National Cancer Institute Center for Cancer Research [NCICCR] data set) available in The Cancer Genome Atlas database. The RNA-seq data were normalized using total reads mapped to the human genome and then normalized using VPS29 as a housekeeping gene, as recommended for RNA-seq data analysis.41 The results showed that BCL6 levels were significantly lower in XABCLs than in NCICCR-DLBCL (P < .005), although low BCL6 mRNA levels were also found in some NCICCR-DLBCL samples (Figure 4E; supplemental Table 6). We tested BCL6 protein levels in the 4 lymphoma cell lines used in this study. BCL6 was readily detectable in Raji cells but not detectable in TC526, JVM2, or Z138, and treatment with MDM2is did not affect the BCL6 level in these cell lines (Figure 4C). This result indicates that expression of BCL6 is inversely associated with MDM2i sensitivity.
Navtemadlin induced tumor regression and prevented systemic dissemination of EBV-positive lymphoma derived from patients with PTLD
To further test the activity of MDM2is in EBV-positive lymphoma in vivo, we evaluated treatment responses of EBV-positive lymphoma cells (TRL1 and TRL595) cultured from tumors from 2 juvenile patients who developed PTLD after bone marrow transplant7 and liver transplant,14 respectively. TRL1 has a 245-bp deletion in the EBV-encoded EBNA3B gene and was resistant to virus-specific adoptive T-cell therapy,7 whereas TRL595 has a frameshift mutation of EBNA3B at amino acid 187.14 In vitro analysis showed that both TRL1 and TRL595 responded to treatment with navtemadlin and idasanutlin, with IC50 ∼0.5 to 1 µM (Figure 5A). Western blot analysis showed that both cell lines were positive for LMP1 but had undetectable BCL6. Treatment with navtemadlin led to upregulation of p53 and p21, as well as cell cycle arrest at G1 and apoptosis (Figure 5B).
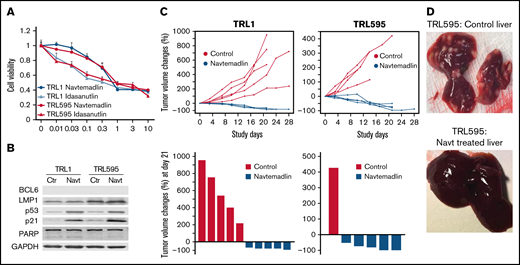
Effect of MDM2is on EBV-positive lymphoma cells derived from patients with PTLD. TRL1 and TRL595 were derived from tumors of patients with PTLD. (A) Dose response to navtemadlin and idasanutlin. Dose-response analysis was performed as described for Figure 4. The data are presented as the mean ± SD of a quadruplicate assay. (B) Protein levels were determined by western blot analysis. Controls were at basal levels; Navt samples were treated with 1 µM navtemadlin for 24 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (C) In vivo treatment response. Animals were enrolled onto the study when tumors reached 200 mm3 and were treated once per day for 21 days. Top: tumor volume changes for each mouse. Bottom: tumor volume changes at day 21 (end of treatment). P < .01 for TRL1. (D) Tumor nodules in liver of TRL595 mice. PARP, poly(ADP-ribose) polymerase.
Effect of MDM2is on EBV-positive lymphoma cells derived from patients with PTLD. TRL1 and TRL595 were derived from tumors of patients with PTLD. (A) Dose response to navtemadlin and idasanutlin. Dose-response analysis was performed as described for Figure 4. The data are presented as the mean ± SD of a quadruplicate assay. (B) Protein levels were determined by western blot analysis. Controls were at basal levels; Navt samples were treated with 1 µM navtemadlin for 24 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (C) In vivo treatment response. Animals were enrolled onto the study when tumors reached 200 mm3 and were treated once per day for 21 days. Top: tumor volume changes for each mouse. Bottom: tumor volume changes at day 21 (end of treatment). P < .01 for TRL1. (D) Tumor nodules in liver of TRL595 mice. PARP, poly(ADP-ribose) polymerase.
To test the in vivo activity of navtemadlin in tumors derived from these 2 cell lines, we established xenograft tumors with TRL1 and TRL595 in NSG mice. The tumor-bearing mice (n = 5 per group) were treated with solvent or navtemadlin when the tumors reached 200 mm3. Four of 5 mice in the control-treated group of TRL595-bearing mice died during days 5 to 14, before the treatment study ended at day 21. Autopsy of these dead mice showed extensive dissemination of B-cell lymphoma in the liver. By the end of treatment at day 21, all surviving control-treated mice had tumor volume increases ranging from ∼200% to 900%. In contrast, all animals treated with navtemadlin survived to the end of the treatment, with tumor regressions ranging from −50% to −89% (Figure 5C).
We euthanized 3 mice per group and the sole surviving mouse in the control-treated group of TRL595-bearing mice and harvested tumor and liver tissue for analysis of the disseminated lymphoma. The results showed massive tumor nodules present in the liver of the mouse bearing TRL595 and treated with solvent, whereas no gross tumor nodules were observed in livers of mice bearing TRL595 and treated with navtemadlin (Figure 5D). We performed immunohistochemical staining with human CD20 antibody to detect systemic dissemination of lymphoma cells in the liver, spleen, lung, and kidney. The entire liver of TRL595-bearing mice treated with solvent was occupied with CD20+ tumor cells, whereas only microscopic tumor nodules (about 1 to 3 per section) were identified in sections of liver from TRL595-bearing mice treated with navtemadlin.
Systemic dissemination of malignant B cells, either as tumor nodules or clusters of tumor cells, were readily identified in spleen, lung, liver, and kidney from TRL595-bearing mice treated with solvent (Figure 6). In contrast, no CD20+ tumor cells were found in sections of spleen, lung, and kidney from TRL595-bearing mice treated with navtemadlin. Systemic dissemination of lymphoma cells was also readily detected in spleen, lung, liver, and kidney in TRL1-bearing mice treated with solvent, but none were seen in TRL1-bearing mice treated with navtemadlin. We tested whether some cell cycle effectors that were downregulated in XABCLs by treatment with MDM2is were also affected by navtemadlin treatment in PTLD models. The results showed that FOXM1, PLK1, and RRM2 were drastically downregulated by navtemadlin treatment in vitro and in vivo (supplemental Figure 2). These results showed that navtemadlin is effective for treating EBV-positive lymphoma derived from tumors of patients with PTLD, inducing molecular changes similar to those observed in XABCLs, and triggering regression and preventing systemic dissemination of established tumors, including those of cancers resistant to adoptive T-cell therapy.
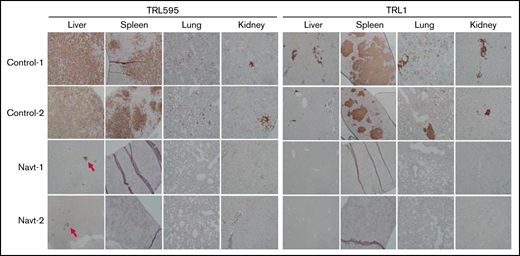
Effect of navtemadlin on systemic dissemination of EBV-positive PTLD lymphoma. Photomicrographs show representative immunohistochemical staining of human CD20 in the liver, spleen, lung, and kidney of 2 control-treated and navtemadlin-treated mice bearing TRL1 and TRL595 tumors. Tumor nodules and clusters of tumor cells were detected in all organs tested in all control mice. No tumor cells were detected in all organs tested in all navtemadlin-treated mice, except that microscopic nodules were observed in liver of TRL595 mice (arrow).
Effect of navtemadlin on systemic dissemination of EBV-positive PTLD lymphoma. Photomicrographs show representative immunohistochemical staining of human CD20 in the liver, spleen, lung, and kidney of 2 control-treated and navtemadlin-treated mice bearing TRL1 and TRL595 tumors. Tumor nodules and clusters of tumor cells were detected in all organs tested in all control mice. No tumor cells were detected in all organs tested in all navtemadlin-treated mice, except that microscopic nodules were observed in liver of TRL595 mice (arrow).
Discussion
The extraordinary response of XABCLs and xenografts of human B-cell lymphoma tumor cells from patients with PTLD to MDM2is in vivo in immunocompromised mice provided proof-of-concept evidence that MDM2-targeted therapy could effectively treat EBV-positive lymphoma in immunocompromised patients. Our results suggest that EBV infection and loss of BCL6 expression could be used as a biomarker to identify responders to MDM2-targeted therapy.
The tumor suppressor gene TP53 is inactivated in about 50% of human cancers by mutations within TP53.42 In cancers with wt TP53, inactivation of p53 frequently occurs through its interaction with its natural inhibitors, such as MDM2.34 MDM2 directly inactivates p53 by blocking its transcriptional activity and by promoting p53 degradation through MDM2 E3 ligase activity.34 MDM2 is also required for survival and growth of some p53-deficient cancer cells, possibly through its interaction with p7343 or residual apoptotic activity of mutant p53.44 Substantial efforts have been undertaken to develop small-molecule inhibitors of the MDM2-p53 interaction for cancer therapy.45 Several of these inhibitors have been tested in clinical trials for cancer therapy,46 including navtemadlin47 and idasanutlin.28 Preclinical studies revealed that navtemadlin induced robust p53 activity, leading to arrest of the cell cycle, apoptosis, inhibition of tumor-cell proliferation, and suppression of tumor growth in vivo in a number of TP53-wt cancer cells but not TP53-mutant cancer cells.48 Knockout of TP53 in the colorectal cancer cell line HCT116 abolished this activity, demonstrating the p53-dependent effects of navtemadlin.48 Clinical studies revealed that navtemadlin had acceptable pharmacokinetics, on-target effects, and promising clinical activity warranting further investigation.47,49 Several studies have also demonstrated that MDM2 is a targetable vulnerability in patients with TP53-wt chronic lymphocytic leukemia and B-cell and T-cell lymphomas.50,51
Previous studies regarding development of targeted therapy have shown that the inability to identify patients who may respond is a major challenge in the early stages of clinical trials of anticancer agents. For example, mutations of the EGFR gene affect response of cancers to treatment with EGFRi52,53 ; notably, neither gefitinib54 nor erlotinib55 was effective in randomized phase 3 trials with unselected patient populations. Similarly, success of the ALK inhibitor crizotinib,56 the BRAF inhibitor vemurafenib,57 and the poly(ADP-ribose) polymerase inhibitor olaparib58 depended on biomarker-based patient stratification in clinical trials. In contrast, a lack of predictive biomarkers for patient stratification curtailed further development of many other investigational drugs, including idasanutlin in a phase 3 trial.59 Thus, efforts have been undertaken to identify gene expression signatures predictive to sensitivity to MDM2 inhibition.60 Our results strongly indicate that EBV infection is a feasible biomarker for vulnerability to MDM2is, and loss of BCL6 expression in tumor cells may also serve as a predictive biomarker for MDM2i vulnerability. BCL6 inhibits p53 expression by direct binding to the target sequence in the TP53 promoter36,37 or by inhibiting p53 protein acetylation,61 and interestingly, BCL6 expression is strongly inhibited in EBV-infected patients with DLBCL.38-40 Among EBV-positive HIV-related non-Hodgkin lymphoma biopsy samples, BCL6 and EBV-encoded LMP1 expression was mutually exclusive.40 BCL6 is generally negative in clinical specimens of EBV-positive DLBCL39 and of EBV-positive DLBCL of the elderly.62 It is likely that EBV-mediated inhibition of BCL6 expression is synthetically lethal to MDM2 inhibition. Indeed, MDM2-dependent inhibition of p53 is required for survival and transformation of EBV-infected B cells.63 However, our efforts to study ectopic expression of BCL6 in B-cell lymphoma cell lines with loss of BCL6 expression have not been successful so far. Thus, this study had some limitations because the causal relationship between loss of BCL6 expression and MDM2i sensitivity is yet to be established. It is not yet clear whether other EBV-positive cancers, such as EBV-associated peripheral T-cell lymphoma, are also susceptible to MDM2is. Nevertheless, our current results support further mechanistic and clinical investigations of MDM2-targeted therapy for EBV-positive lymphoma.
Acknowledgments
The authors thank Sarah Bronson, ELS, and the Research Medical Library at The University of Texas MD Anderson Cancer Center for editorial review of the manuscript.
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute (R01 CA190628), The University of Texas PDX Development and Trial Center (U54 CA224065), Specialized Program of Research Excellence (SPORE) (CA070907), Experimental Therapeutics Clinical Trials Network (UM1CA186688), and The University of Texas Anderson Cancer Center (P30 CA016672), and by funds from the University Cancer Foundation via the Lung Cancer Moon Shot Program.
We used Advanced Technology Genomics Core, Bioinformatics Shared Resource, Biostatistics Resource Group, Flow Cytometry and Cellular Imaging Core, Functional Proteomics Reverse Phase Protein Array Core, Characterized Cell Line Core (now Cytogenetics and Cell Authentication Core), Tissue Biospecimen and Pathology Resource, and the Research Animal Support Facility.
Authorship
Contribution: B.F. designed and directed the study and wrote the first draft of the manuscript; X.Z., R.Z., C.R., S.W., Y.X., A.P., S.W., Y.Y., and H.H. performed experiments, analyzed data, and helped write the manuscript; X.S., C.M., H.C., W.M., J.W., and J.Z. analyzed and interpreted the data; C.M.R. provided patient-derived tumor cells from patients with PTLD; and F.M.-B., R.E.C., C.M.R., J.V.H., S.G.S., A.A.V., J.A.R., M.J.Y., and M.W. participated in study design, data interpretation, and writing the manuscript.
Conflict-of-interest disclosure: J.A.R. received a commercial research grant from MDACC, has a sponsored research agreement from Genprex, has ownership interest (including stock and patents) in Genprex, and is a consultant/advisory board member for Genprex. F.M.-B. has served as a consultant for Aduro BioTech, Debiopharm, eFFECTOR Therapeutics, F. Hoffmann-La Roche, Genentech, IBM Watson, The Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer, Samsung Bioepis, Seattle Genetics, Tyra Biosciences, Xencor, and Zymeworks; has served on an advisory committee for Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology, Seattle Genetics, Silverback Therapeutics, and Spectrum Pharmaceuticals; has received research sponsorship funding from Aileron Therapeutics, AstraZeneca, Bayer Healthcare Pharmaceuticals, Calithera Biosciences, Curis, CytomX Therapeutics, Daiichi Sankyo, Debiopharm International, eFFECTOR Therapeutics, Genentech, Guardant Health, Millennium Pharmaceuticals, Novartis, Puma Biotechnology, and Taiho Pharmaceuticals; has received honoraria from Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey, and the University of Texas Health San Antonio. C.M.R. and spouse are founding members of Allogene and Marker Therapeutics, both have significant financial interest with Bellicum Pharmaceuticals and Tessa Therapeutics; and the spouse has significant conflicts of interest with Abintus, Bluebird Bio, Memgen, TScan, Turnstone Biologics, and Walking Fish. M.W. has served as a consultant for AstraZeneca, Bayer Healthcare, BeiGene, CSTone, DTRM Biopharma (Cayman), Epizyme, Genentech, InnoCare, Janssen, Juno, Kite Pharma, Loxo Oncology, Miltenyi Biomedicine, Oncternal, Pharmacyclics, and VelosBio; has received research funding from Acerta Pharma, AstraZeneca, BeiGene, BioInvent, Celgene, Genentech, Innocare, Janssen, Juno, Kite Pharma, Eli Lilly, Loxo Oncology, Molecular Templates, Oncternal, Pharmacyclics, and VelosBio; and has received honoraria from Acerta Pharma, Anticancer Association, AstraZeneca, BeiGene, BGICS, CAHON, Chinese Medical Association, Clinical Care Options, Dava Oncology, Eastern Virginia Medical School, Epizyme, Hebei Cancer, Prevention Federation, Imedex, Janssen, Kite Pharma, TS Oncology, Miltenyi Biomedicine, Moffit Cancer Center, Mumbai Hematology Group, Newbridge Pharmaceuticals, OMI, Pharmacyclics, Physicians Education, Resources (PER), Scripps, and The First Afflicted Hospital of Zhejiang University. The remaining authors declare no competing financial interests.
Correspondence: Bingliang Fang, Department of Thoracic and Cardiovascular Surgery, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 1489, Houston, TX 77030; e-mail: bfang@mdanderson.org.

