

Key Points
Inotuzumab was an effective treatment of B-ALL relapse post-blinatumomab regardless of CD19 expression status.
Outcomes of R/R B-ALL patients relapsing after treatment with both blinatumomab and inotuzumab were poor, with high relapse and mortality.

Abstract
Novel monoclonal antibody (mAb)-based therapies targeting CD19 and CD22 (blinatumomab and inotuzumab) have shown high rates of complete remission (CR) and been used as a bridging treatment to potentially curative allogeneic hematopoietic stem cell transplantation (alloHSCT) in adults with relapsed or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL). However, limited data exist on the outcome of patients resistant to both mAbs as well as responses to each agent when progressed after the alternate antigen-targeted mAb. Herein, we report outcomes of 29 patients with R/R B-ALL previously treated with both blinatumomab and inotuzumab. Twenty-five patients (86.2%) received blinatumomab as first mAb (mAb1), and CD19-negative/dim relapses were observed in 44% of the patients. Inotuzumab induced CR in 68% of the patients for post-blinatumomab relapse regardless of CD19 expression status. The median time between mAb1 and mAb2 was 99 days. Twelve (63.2%) of 19 patients who achieved remission after mAb2 underwent alloHSCT. The median time from mAb2 to alloHSCT was 37.5 days. Acute graft-versus-host disease and nonrelapse mortality were observed in 58.3% (grade 3 or higher, 25%) and 41.7%, respectively. With a median follow-up of 16.8 months after mAb2, 19 patients (65.5%) relapsed, and 21 patients (72.4%) have died. Overall survival was not different between alloHSCT and non-alloHSCT patients. In conclusion, patients with B-ALL who relapsed after blinatumomab could be successfully rescued by inotuzumab as a bridge to alloHSCT but represent an ultra-high-risk group with poor overall survival. Further studies, including novel consolidation and treatment sequence, may improve outcomes of these patients.
Introduction
The prognosis of patients with relapsed or refractory (R/R) precursor B-cell acute lymphoblastic leukemia (B-ALL) has historically been dismal.1,2 Allogeneic hematopoietic stem cell transplantation (alloHSCT) has been the major approach used with potential to provide long-term remission for these high-risk patients.3 Historically, however, only a small proportion of patients with R/R B-ALL have eventually undergone alloHSCT, mostly due to inability to attain disease control or because of compromised organ function.2 More recently, monoclonal antibody (mAb)-based treatments targeting CD19 and CD22 have become more widely used modalities for the treatment of patients with R/R B-ALL. Blinatumomab, a CD3/CD19–targeted bispecific T-cell engager consisting of 2 linked single-chain variable fragments, and inotuzumab ozogamicin (inotuzumab), an antibody–drug conjugate comprising a humanized anti-CD22 mAb linked to a calicheamicin toxin, have shown superior antileukemic activity compared with conventional chemotherapy and have become preferred salvage treatment strategies, including as a bridge to alloHSCT, in patients with R/R B-ALL.4-6 The majority of published clinical data of blinatumomab and inotuzumab are from clinical trials that predominantly reported initial responses to either of these agents only, whereas clinical course and outcomes of patients who received both mAbs have not been well described. Limited data are available on whether patients who relapse after blinatumomab or inotuzumab derive any therapeutic benefit to the alternate mAb.7 Moreover, the clinical benefit and safety of alloHSCT after both blinatumomab and inotuzumab in these heavily treated patients are unclear.
Given the routine use of blinatumomab and inotuzumab in clinical practice, patients who relapse after or develop resistance to both of these agents will become increasingly more common. We therefore studied treatment outcomes and toxicities in 29 patients with B-ALL who received both blinatumomab and inotuzumab for relapsed diseases at our institution. We report patient and disease characteristics, patterns of relapse after each mAb with regard to antigen expression, response to the alternate mAb, and survival with and without subsequent alloHSCT.
Patients and methods
We reviewed the patient charts of 29 patients aged ≥15 years with R/R B-ALL who had received salvage therapy for morphologic or measurable (minimal) residual disease (MRD) with both blinatumomab and inotuzumab at Memorial Sloan Kettering Cancer Center between January 2012 and December 2019. Baseline characteristics, details of mAb therapy (including the sequence and schedule of mAb), previous alloHSCT, previous chimeric antigen receptor (CAR) T-cell therapy, interim treatments between 2 mAbs, and post-mAb treatments were extracted from the electronic health record of each patient. Blinatumomab and inotuzumab were administered following the standard dosing and schedules as approved by the US Food and Drug Administration. Response, outcome, and treatment-related adverse events after mAb administration were described. The study protocol was reviewed and approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board and conducted in accordance with the Declaration of Helsinki. The cutoff date for data analysis was June 30, 2020.
Complete remission (CR) was defined as bone marrow lymphoblasts <5% and absence or resolution of extramedullary leukemic foci, with or without hematopoietic recovery. MRD was assessed in bone marrow aspirate samples by using multiparameter flow cytometry with sensitivity of at least 10−4 of total leukocyte events as described previously.8 Loss or lack of expression of CD19 and CD22 was defined as follows: samples were generally described as “negative” for expression if <20% of the abnormal cells showed positivity above the level of the internal negative controls, whereas dim expression was generally defined as median expression of at least one-third of a log (∼3-fold) decrease of expression compared with expected expression by normal immature B cells on standardized instruments. Based on the expression density on flow cytogram and clinical implication of target antigen density, we classify the expression status of CD19 and CD22 into 2 groups: negative or low (neg/low) and positive (pos) antigen as previously described.9,10
In patients who underwent alloHSCT after mAb salvage therapy, transplant-associated parameters, including donor–patient characteristics, conditioning regimens, graft-versus-host disease (GVHD) prophylaxis, outcome, and complications, were reported. Acute GVHD (aGVHD) was staged and graded according to the 1994 Consensus Conference on aGVHD Grading system.11
Cytokine release syndrome (CRS) and neurotoxicities after blinatumomab were diagnosed and re-classified based on the American Society of Transplant and Cellular Therapy consensus guidelines.12 Hepatic sinusoidal obstructive syndrome (SOS) was defined based on revised classification from the European Society for Blood and Marrow Transplantation,13 which includes bilirubin ≥2 mg/dL and 2 criteria of painful hepatomegaly, weight gain >5%, and ascites.
Event-free survival (EFS) was defined as the time from mAb treatment initiation to leukemic progression or death, whichever occurred first. Overall survival (OS) was the duration between the mAb treatment to death from any causes. Cumulative incidence of relapse/progression was the proportion of relapse/progression after mAb administration, with the competing risk being death from nonrelapse etiologies.
Statistical analysis
Continuous variables are described by using median and range. Categorical variables are reported in number and percentage. Comparison between groups was performed by using the Kruskal-Willis rank sum test for continuous variables and the χ2 or Fisher’s exact test for categorical variables. The EFS and OS were estimated by using the Kaplan-Meier analysis and compared between groups by using the log-rank test. Cumulative incidence was analyzed by competing risk analysis as described in the Fine and Gray subdistribution hazard function. A P value <.05 was considered statistically significance. Analyses were performed by using R software version 4.0.2 (R Foundation for Statistical Computing).
Results
Patient characteristics and sequence of mAb therapy
Table 1 describes the baseline characteristics of 29 patients who received both blinatumomab and inotuzumab for R/R B-ALL in this study. The consort diagram of treatment sequence and response of 29 patients is shown in Figure 1. Twenty-five (86.2%) patients received blinatumomab as their first mAb (mAb1) and inotuzumab as the second agent (mAb2) representing the majority of patients in the study cohort; the other 4 patients (13.8%) had inotuzumab as their mAb1.
Baseline characteristics of 29 patients who received both blinatumomab and inotuzumab ozogamicin for R/R precursor R/R as a whole cohort and as stratified according to the sequence of blinatumomab and inotuzumab ozogamicin
| Characteristic | All patients (N = 29) | Blinatumomab mAb1 (n = 25) | Inotuzumab mAb1 (n = 4) |
|---|---|---|---|
| Median age at diagnosis (IQR, y) | 45.3 (25.1-62.6) | 43.6 (24.4-60.7) | 61.2 (54.6-70.6) |
| Male sex | 17 (58.6) | 13 (52.0) | 4 (100.0) |
| Cytogenetic stratification* | |||
| High risk | 12 (41.4) | 10 (40.0) | 2 (50.0) |
| Standard risk | 14 (48.3) | 12 (48.0) | 2 (50.0) |
| Missing data | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Philadelphia chromosome–positive B-ALL | 4 (13.8) | 4 (16.0) | 0 (0.0) |
| Presence of extramedullary disease | 8 (27.6) | 6 (24.0) | 2 (50.0) |
| Complete response to first-line induction therapy | |||
| Relapse after achieving CR | 24 (82.8) | 22 (88.0) | 2 (50.0) |
| Refractory | 5 (17.2) | 3 (12.0) | 2 (50.0) |
| Median no. of prior treatment lines before first mAb (range, lines) | 1 (1-5) | 1 (1-5) | 1 (1-2) |
| Prior alloHSCT before mAb1 | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Prior CD19 CAR T-cell therapy before mAb1 | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Median bone marrow blast percentage at mAb1 (IQR, %) | 9 (3-25) | 9 (3-20) | 25 (14-58) |
| MRD at the time of mAb1 | 7 (24.1) | 7 (28.0) | 0 (0.0) |
| Median time from mAb1 to mAb2 (IQR, d) | 99 (35-240) | 99 (35-240) | 116 (55-221) |
| Received interim treatment between 2 mAbs | 14 (48.3) | 13 (52.0) | 1 (25.0) |
| Median number of blinatumomab cycles (range, cycles) | 1 (1-6) | 1 (1-6) | 2 (1-5) |
| Median number of inotuzumab cycles (range, cycles) | 2 (1-5) | 2 (1-5) | 2 (1-2) |
| alloHSCT after mAb2 | 12 (41.4) | 11 (44.0) | 1 (25.0) |
| Characteristic | All patients (N = 29) | Blinatumomab mAb1 (n = 25) | Inotuzumab mAb1 (n = 4) |
|---|---|---|---|
| Median age at diagnosis (IQR, y) | 45.3 (25.1-62.6) | 43.6 (24.4-60.7) | 61.2 (54.6-70.6) |
| Male sex | 17 (58.6) | 13 (52.0) | 4 (100.0) |
| Cytogenetic stratification* | |||
| High risk | 12 (41.4) | 10 (40.0) | 2 (50.0) |
| Standard risk | 14 (48.3) | 12 (48.0) | 2 (50.0) |
| Missing data | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Philadelphia chromosome–positive B-ALL | 4 (13.8) | 4 (16.0) | 0 (0.0) |
| Presence of extramedullary disease | 8 (27.6) | 6 (24.0) | 2 (50.0) |
| Complete response to first-line induction therapy | |||
| Relapse after achieving CR | 24 (82.8) | 22 (88.0) | 2 (50.0) |
| Refractory | 5 (17.2) | 3 (12.0) | 2 (50.0) |
| Median no. of prior treatment lines before first mAb (range, lines) | 1 (1-5) | 1 (1-5) | 1 (1-2) |
| Prior alloHSCT before mAb1 | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Prior CD19 CAR T-cell therapy before mAb1 | 3 (10.3) | 3 (12.0) | 0 (0.0) |
| Median bone marrow blast percentage at mAb1 (IQR, %) | 9 (3-25) | 9 (3-20) | 25 (14-58) |
| MRD at the time of mAb1 | 7 (24.1) | 7 (28.0) | 0 (0.0) |
| Median time from mAb1 to mAb2 (IQR, d) | 99 (35-240) | 99 (35-240) | 116 (55-221) |
| Received interim treatment between 2 mAbs | 14 (48.3) | 13 (52.0) | 1 (25.0) |
| Median number of blinatumomab cycles (range, cycles) | 1 (1-6) | 1 (1-6) | 2 (1-5) |
| Median number of inotuzumab cycles (range, cycles) | 2 (1-5) | 2 (1-5) | 2 (1-2) |
| alloHSCT after mAb2 | 12 (41.4) | 11 (44.0) | 1 (25.0) |
Data are presented as no. (%) unless otherwise indicated.
Based on UKALLXII/ECOG2993.2
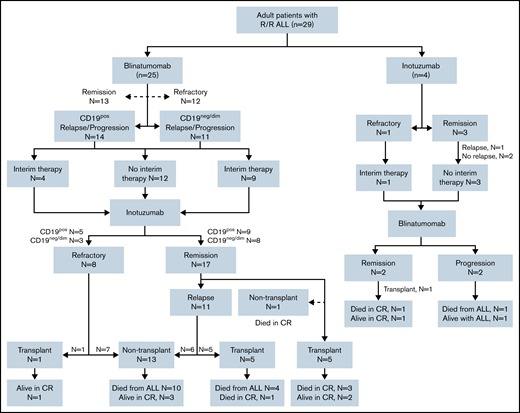
Consort diagram delineates treatment distribution of the entire cohort of 29 patients. Twenty-five patients received blinatumomab as the mAb1 and inotuzumab as the mAb2. Four patients received inotuzumab mAb1 and blinatumomab mAb2.
Consort diagram delineates treatment distribution of the entire cohort of 29 patients. Twenty-five patients received blinatumomab as the mAb1 and inotuzumab as the mAb2. Four patients received inotuzumab mAb1 and blinatumomab mAb2.
Sixteen (55.2%) of 29 patients attained morphologic CR after mAb1 (MRDneg CR in 10 [34.4%]): 13 of 25 patients after blinatumomab, and 3 of 4 patients after inotuzumab. Nineteen patients (65.5%) achieved CR after mAb2: 17 of 25 patients after inotuzumab mAb2, and 2 of 4 patients after blinatumomab mAb2. The median time between mAb1 and mAb2 was 99 days (interquartile range [IQR], 35-240 days). Twelve patients (41.4%) received interim non–mAb-based salvage treatments before mAb2. Overall, 12 patients (41.4%) underwent alloHSCT after mAb2, and 2 received CD19 CAR T cells after mAb2 (1 patient received CAR T-cell therapy followed by alloHSCT).
Patients who received mAb1 with blinatumomab (blinatumomab mAb1)
Of the 25 patients who received blinatumomab as mAb1, 7 (28.0%) had MRD, and 18 (72.0%) had morphologic disease at the time of blinatumomab mAb1 therapy. Four patients with Philadelphia chromosome–positive ALL were on ponatinib at the time of relapse and continued ponatinib concurrently with blinatumomab mAb1. The median number of blinatumomab cycle was 1 (range, 1-6). Thirteen patients (52.0%) achieved CR or complete remission with incomplete count recovery, including MRDneg CR in 8 patients. CRS and immune effector cell–associated neurotoxicity syndrome of all grades were observed in 52.0% and 28.0%, respectively. At the time of relapse/progression after blinatumomab, 11 patients (44.0%) progressed with CD19 negative/dim (CD19neg/dim) disease, whereas 14 patients (56.0%) had persistent CD19 expression (CD19pos). There was no difference in median time from blinatumomab mAb1 to relapse/progression between CD19pos and CD19neg/dim relapse (65 vs 73 days; P = .74). Of the 25 patients who received blinatumomab mAb1, 6 (24%) underwent alloHSCT after blinatumomab mAb1, and all subsequently relapsed before receiving inotuzumab mAb2 (supplemental Table 1). The median number of inotuzumab mAb2 cycles was 2 (range, 1-5). The median total administered dose of inotuzumab mAb2 was 1.8 mg/m2 (range, 1.8-3.6 mg/m2). Three patients (12.0%) received combined chemotherapy with inotuzumab mAb2 (mini-hyper-CVD: cyclophosphamide, vincristine, doxorubicin in 2 patients and vincristine in 1 patient). In 4 Philadelphia chromosome–positive patients with ALL, ponatinib was discontinued and was not given with inotuzumab mAb2.
The CR rate after inotuzumab mAb2 was 68.0% (17 of 25; MRDneg CR, 48.0%). The response rates to inotuzumab mAb2 did not differ between CD19neg/dim and CD19pos relapse post-blinatumomab; 8 of 11 CD19neg/dim relapse patients achieved CR (4 MRDneg and 4 MRDpos CR) vs 9 of 14 CD19pos relapse patients (8 MRDneg and 1 MRDpos CR). Supplemental Table 2 summarizes patient characteristics, management, and outcomes stratified according to CD19 expression at relapse after blinatumomab mAb1. Thirteen patients (52.0%) underwent non–mAb-based interim therapies for post-blinatumomab progression before receiving inotuzumab mAb2. The median number of interim salvage therapies between blinatumomab mAb1 and inotuzumab mAb2 was 2 (range, 1-5), and the median time from blinatumomab mAb1 to inotuzumab mAb2 was 99 days (141 days in CD19neg/dim and 52 days in CD19pos; P = .11). There was no significant difference in CR rates to inotuzumab mAb2 between patients who received non-mAb salvage therapies before inotuzumab and patients who proceeded directly to inotuzumab (69.2% vs 66.7%; P = .89) (supplemental Figure 1). Table 2 delineates the characteristics, management, and outcomes of patients receiving blinatumomab mAb1 as stratified according to whether the patients had interim salvage therapy before inotuzumab mAb2.
Characteristics of 25 patients who received blinatumomab mAb1 and inotuzumab mAb2 stratified according to the receipt of other interim therapy between mAb1 and mAb2
| Characteristic | All (N = 25) | No interim therapy (n = 12) | Received interim therapy (n = 13) | P |
|---|---|---|---|---|
| Median age at diagnosis (IQR, y) | 43.6 (24.4-60.7) | 42.0 (21.2-65.0) | 43.6 (32.4-47.6) | .46 |
| Age >45 y | 12 (48.0%) | 6 (50.0%) | 6 (46.2%) | .84 |
| Male sex | 13 (52.0%) | 5 (41.7%) | 8 (61.5%) | .32 |
| Median number of prior lines of therapy before blinatumomab (range, lines) | 1 (1-5) | 2 (1-3) | 1 (1-5) | .04 |
| Prior alloHSCT before mAb1 | 3 (12.0%) | 1 (8.3%) | 2 (15.4%) | 1.00 |
| High-risk cytogenetic abnormalities | 10 (40.0%) | 4 (33.3%) | 6 (46.2%) | .21 |
| Philadelphia chromosome positive | 4 (16.0%) | 3 (25.0%) | 1 (7.1%) | .32 |
| Presence of MLL rearrangement | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | 1.00 |
| Disease burden at the time of blinatumomab mAb1 | .59 | |||
| MRD | 7 (28.0%) | 4 (33.3%) | 3 (23.1%) | |
| Morphologic disease | 18 (72.0%) | 8 (66.7%) | 10 (76.9%) | |
| Site of disease at the time of blinatumomab mAb1 | .78 | |||
| Isolated marrow | 21 (84.0%) | 10 (83.3%) | 11 (84.6%) | |
| Isolated extramedullary | 1 (4.0%) | 0 (0.0%) | 1 (7.7%) | |
| Both | 3 (12.0%) | 2 (16.7%) | 1 (7.7%) | |
| Median no. of blinatumomab mAb1 cycles (range, cycles) | 1 (1-6) | 1 (1-6) | 1 (1-4) | .53 |
| Best response to blinatumomab mAb1 | .32 | |||
| CR | 13 (52.0%) | 5 (41.7%) | 8 (61.5%) | |
| No response | 12 (48.0%) | 7 (58.3%) | 5 (38.5%) | |
| Median time from blinatumomab to progression/relapse (IQR, d) | 73 (34-219) | 65 (37-118) | 73 (33-290) | .85 |
| Site of disease at the time of relapse following blinatumomab mAb1 | .41 | |||
| Isolated marrow | 15 (60.0%) | 8 (66.7%) | 7 (53.8%) | |
| Isolated extramedullary | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | |
| Both | 9 (36.0%) | 3 (25.0%) | 6 (46.2%) | |
| Central nervous system involvement at the time of relapse after blinatumomab mAb1 | 1 (4.0%) | 0 (0.0%) | 1 (7.7%) | .48 |
| Response to inotuzumab mAb2 | .89 | |||
| CR | 17 (68.0%) | 8 (69.2%) | 9 (69.2%) | |
| No response | 8 (32.0%) | 4 (30.8%) | 4 (30.8%) |
| Characteristic | All (N = 25) | No interim therapy (n = 12) | Received interim therapy (n = 13) | P |
|---|---|---|---|---|
| Median age at diagnosis (IQR, y) | 43.6 (24.4-60.7) | 42.0 (21.2-65.0) | 43.6 (32.4-47.6) | .46 |
| Age >45 y | 12 (48.0%) | 6 (50.0%) | 6 (46.2%) | .84 |
| Male sex | 13 (52.0%) | 5 (41.7%) | 8 (61.5%) | .32 |
| Median number of prior lines of therapy before blinatumomab (range, lines) | 1 (1-5) | 2 (1-3) | 1 (1-5) | .04 |
| Prior alloHSCT before mAb1 | 3 (12.0%) | 1 (8.3%) | 2 (15.4%) | 1.00 |
| High-risk cytogenetic abnormalities | 10 (40.0%) | 4 (33.3%) | 6 (46.2%) | .21 |
| Philadelphia chromosome positive | 4 (16.0%) | 3 (25.0%) | 1 (7.1%) | .32 |
| Presence of MLL rearrangement | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | 1.00 |
| Disease burden at the time of blinatumomab mAb1 | .59 | |||
| MRD | 7 (28.0%) | 4 (33.3%) | 3 (23.1%) | |
| Morphologic disease | 18 (72.0%) | 8 (66.7%) | 10 (76.9%) | |
| Site of disease at the time of blinatumomab mAb1 | .78 | |||
| Isolated marrow | 21 (84.0%) | 10 (83.3%) | 11 (84.6%) | |
| Isolated extramedullary | 1 (4.0%) | 0 (0.0%) | 1 (7.7%) | |
| Both | 3 (12.0%) | 2 (16.7%) | 1 (7.7%) | |
| Median no. of blinatumomab mAb1 cycles (range, cycles) | 1 (1-6) | 1 (1-6) | 1 (1-4) | .53 |
| Best response to blinatumomab mAb1 | .32 | |||
| CR | 13 (52.0%) | 5 (41.7%) | 8 (61.5%) | |
| No response | 12 (48.0%) | 7 (58.3%) | 5 (38.5%) | |
| Median time from blinatumomab to progression/relapse (IQR, d) | 73 (34-219) | 65 (37-118) | 73 (33-290) | .85 |
| Site of disease at the time of relapse following blinatumomab mAb1 | .41 | |||
| Isolated marrow | 15 (60.0%) | 8 (66.7%) | 7 (53.8%) | |
| Isolated extramedullary | 1 (4.0%) | 1 (8.3%) | 0 (0.0%) | |
| Both | 9 (36.0%) | 3 (25.0%) | 6 (46.2%) | |
| Central nervous system involvement at the time of relapse after blinatumomab mAb1 | 1 (4.0%) | 0 (0.0%) | 1 (7.7%) | .48 |
| Response to inotuzumab mAb2 | .89 | |||
| CR | 17 (68.0%) | 8 (69.2%) | 9 (69.2%) | |
| No response | 8 (32.0%) | 4 (30.8%) | 4 (30.8%) |
MLL, mixed lymphoid lineage.
Of 17 patients who re-achieved CR after inotuzumab mAb2, 10 (58.8%) underwent consolidative alloHSCT. Hepatotoxicity was seen in 14 patients during/after inotuzumab mAb2 (transaminitis any grade, n = 12; hyperbilirubinemia any grade, n = 6). Four patients fulfilled the diagnostic criteria of hepatic SOS, all of which occurred after alloHSCT (three with grade 3, and one with grade 5). At the time of data cutoff, 19 (76%) of 25 patients who received blinatumomab mAb1 have died (14 from progressive disease and 5 from treatment-related mortality), including 10 of CD19neg/dim and 9 of CD19pos disease. At the time of relapse, CD22 expression data were available in 15 (60%) of 25 patients after inotuzumab mAb2 (CD22pos, n = 8; CD22neg/dim, n = 7). The details of CD19 and CD22 status during disease course in relation to mAbs therapy are shown in supplemental Table 3.
Patients who received mAb1 with inotuzumab ozogamicin (inotuzumab mAb1)
Among 4 patients who received inotuzumab as the mAb1, three achieved CR and one had persistent disease. All 4 patients received inotuzumab mAb1 as a monotherapy and received blinatumomab mAb2 immediately after inotuzumab. Of the 3 patients who achieved CR from inotuzumab mAb1, one received blinatumomab mAb2 as a bridge to alloHSCT, and the other 2 received blinatumomab mAb2 as a consolidation, but all subsequently relapsed. One patient who had persistent diseases after inotuzumab mAb1 was refractory to salvage blinatumomab mAb2. The median time between inotuzumab mAb1 and blinatumomab mAb2 was 116 days (range, 17-387 days). All 4 patients had persistent CD22 and CD19 expression at the time of relapse after both inotuzumab and blinatumomab. No patient developed hepatotoxicity or hepatic SOS after inotuzumab mAb1, whereas 1 patient developed grade 1 CRS and grade 1 immune effector cell–associated neurotoxicity syndrome after blinatumomab mAb2. At the time of data cutoff, 2 patients have died (1 from progressive disease, 1 from treatment-related toxicity), 1 remains alive in CR after inotuzumab re-treatment, and 1 remains alive with persistent disease after receiving non–mAb-based salvage treatments.
Patients who underwent alloHSCT after mAb2 therapy
Twelve patients underwent alloHSCT after mAb2: 10 patients post-inotuzumab mAb2, 1 patient post-blinatumomab mAb2, and 1 patient after CD19 CAR T-cell therapy post-inotuzumab mAb2. All patients were in CR at the time of alloHSCT. Of these 12 alloHSCT patients, 1 had undergone prior alloHSCT. Table 3 summarizes detailed transplant characteristics of 12 alloHSCT patients. Seven had alloHSCT with myeloablative conditioning regimens and 4 received ex vivo T cell–depleted grafts. Neutrophil engraftment was observed in 11 patients (91.7%) at the median time of 12 days (IQR, 11-24 days) from stem cell infusion. Seven patients (58.3%) developed aGVHD (grade 3 or higher in 3 patients [stage 3 lower gastrointestinal, n = 1; stage 2 lower gastrointestinal, n = 1; and stage 3 liver, n = 1]). aGVHD was observed in 50% of patients who had a T cell–depleted graft and 71% with unmanipulated graft. Hepatic SOS was observed in 4 patients, all of whom received inotuzumab as the mAb2, with a median time from inotuzumab mAb2 to alloHSCT of 59 days (range, 49-116 days) and underwent transplant with myeloablative conditioning regimens (clofarabine/thiotepa/melphalan, n = 3; fludarabine/cyclophosphamide/thiotepa, n = 1). The details of the 12 patients who underwent alloHSCT after mAb2 are included in supplemental Table 4.
Detailed information of 12 patients who underwent alloHSCT after mAb2
| Characteristic | N = 12 |
|---|---|
| Median age at transplantation (IQR, y) | 49 (17-42) |
| Disease status at the time of transplantation | |
| MRDnegative CR | 9 (75.0) |
| MRDpositive CR | 3 (25.0) |
| Graft source | |
| Bone marrow | 2 (16.7) |
| Peripheral blood | 8 (66.7) |
| Cord blood | 2 (16.7) |
| Donor | |
| Related | 3 (25.0) |
| HLA matched sibling | 1 (8.3) |
| HLA haploidentical | 2 (16.7) |
| Unrelated | 7 (58.3) |
| Umbilical cord blood or haploidentical-umbilical cord | 2 (16.7) |
| Recipient–donor gender relation | |
| Male–male | 3 (25.0) |
| Male–female | 2 (16.7) |
| Female–male | 3 (25.0) |
| Female–female | 3 (25.0) |
| NA* | 1 (8.3) |
| Histocompatibility | |
| 10/10 (fully HLA matched) | 6 (50.0) |
| 9/10 (one HLA locus mismatched) | 2 (16.7) |
| ≤8/10 (2 or more loci mismatched including haploidentical HLA) | 4 (33.3) |
| Conditioning regimens | |
| Myeloablative | |
| Clofarabine, busulfan 16 mg/kg | 2 (16.7) |
| Cyclophosphamide, thiotepa, TBI | 2 (16.7) |
| Fludarabine, cyclophosphamide, thiotepa, TBI | 1 (8.3) |
| Clofarabine, melphalan, thiotepa | 2 (16.7) |
| Reduced intensity or non-myeloablative | |
| Fludarabine, cyclophosphamide, TBI-200 | 2 (16.7) |
| Fludarabine, melphalan | 3 (25.0) |
| GVHD prophylaxis | |
| Ex vivo T-cell depletion (CD34 selection) | 5 (41.7) |
| Posttransplant cyclophosphamide | 3 (25.0) |
| Methotrexate, calcineurin inhibitor | 3 (25.0) |
| Calcineurin inhibitor, mycophenolate mofetil | 1 (8.3) |
| Median number of CD34+ dose (IQR, ×106 cells/kg) | 5.02 (4.7-7.4) |
| Median time from infusion to neutrophil engraftment (IQR, d) | 12 (11-24) |
| GVHD | 6/12 (50) |
| Grade 1 | 1/6 (16.7) |
| Grade 2 | 2/6 (33.3) |
| Grade 3 | 1/6 (16.7) |
| Grade 4 | 2/6 (33.3) |
| SOS | 4 (33.3) |
| Relapse after alloHSCT | 5 (41.7) |
| Death after alloHSCT | 9 (75.0) |
| Died of progressive disease | 4/9 (44.4) |
| Died in remission | 5/9 (55.6) |
| Characteristic | N = 12 |
|---|---|
| Median age at transplantation (IQR, y) | 49 (17-42) |
| Disease status at the time of transplantation | |
| MRDnegative CR | 9 (75.0) |
| MRDpositive CR | 3 (25.0) |
| Graft source | |
| Bone marrow | 2 (16.7) |
| Peripheral blood | 8 (66.7) |
| Cord blood | 2 (16.7) |
| Donor | |
| Related | 3 (25.0) |
| HLA matched sibling | 1 (8.3) |
| HLA haploidentical | 2 (16.7) |
| Unrelated | 7 (58.3) |
| Umbilical cord blood or haploidentical-umbilical cord | 2 (16.7) |
| Recipient–donor gender relation | |
| Male–male | 3 (25.0) |
| Male–female | 2 (16.7) |
| Female–male | 3 (25.0) |
| Female–female | 3 (25.0) |
| NA* | 1 (8.3) |
| Histocompatibility | |
| 10/10 (fully HLA matched) | 6 (50.0) |
| 9/10 (one HLA locus mismatched) | 2 (16.7) |
| ≤8/10 (2 or more loci mismatched including haploidentical HLA) | 4 (33.3) |
| Conditioning regimens | |
| Myeloablative | |
| Clofarabine, busulfan 16 mg/kg | 2 (16.7) |
| Cyclophosphamide, thiotepa, TBI | 2 (16.7) |
| Fludarabine, cyclophosphamide, thiotepa, TBI | 1 (8.3) |
| Clofarabine, melphalan, thiotepa | 2 (16.7) |
| Reduced intensity or non-myeloablative | |
| Fludarabine, cyclophosphamide, TBI-200 | 2 (16.7) |
| Fludarabine, melphalan | 3 (25.0) |
| GVHD prophylaxis | |
| Ex vivo T-cell depletion (CD34 selection) | 5 (41.7) |
| Posttransplant cyclophosphamide | 3 (25.0) |
| Methotrexate, calcineurin inhibitor | 3 (25.0) |
| Calcineurin inhibitor, mycophenolate mofetil | 1 (8.3) |
| Median number of CD34+ dose (IQR, ×106 cells/kg) | 5.02 (4.7-7.4) |
| Median time from infusion to neutrophil engraftment (IQR, d) | 12 (11-24) |
| GVHD | 6/12 (50) |
| Grade 1 | 1/6 (16.7) |
| Grade 2 | 2/6 (33.3) |
| Grade 3 | 1/6 (16.7) |
| Grade 4 | 2/6 (33.3) |
| SOS | 4 (33.3) |
| Relapse after alloHSCT | 5 (41.7) |
| Death after alloHSCT | 9 (75.0) |
| Died of progressive disease | 4/9 (44.4) |
| Died in remission | 5/9 (55.6) |
Data are presented as no. (%) unless otherwise indicated.
NA, not applicable; TBI, total body irradiation.
Patients who received single cord blood transplantation, HLA match 4/6, no available data on sex of the cord unit.
Patients who did not undergo alloHSCT after mAb2 therapy
Seventeen patients did not undergo alloHSCT after mAb2 because of active disease (n = 12), patient or physician preference despite maintaining CR (n = 4), or salvage treatment–related mortality (n = 1). Thirteen (76.5%) of 17 patients received additional salvage therapies after mAb2. The median number of salvage treatments after relapse post-mAb2 in nontransplant patients was 2 lines (range, 1-3), including CD19 CAR T-cell therapy in 2 patients and mAb retreatment in 5 patients (3 with blinatumomab and 2 with inotuzumab). The median time from the mAb2 to the non-transplant salvage therapy was 100 days (IQR, 61-198 days). The 2 patients who received CD19 CAR T-cell therapy for post-mAb2 relapse did not respond to the treatment. Among the 5 patients who were re-treated with blinatumomab and inotuzumab, 1 patient who received inotuzumab mAb1 achieved CR to inotuzumab retreatment for relapse following blinatumomab mAb2 and remains alive in continuous CR at the time of last follow-up; 1 patient who received blinatumomab mAb1 achieved CR to subsequent blinatumomab retreatment but remission was short-lived (3 months).
Survival outcomes
At a median follow-up duration of 16.8 months, 21 patients (72.4%) had died. The corresponding 1-year OS after mAb2 was 33.1% (95% confidence interval [CI], 19.4-36.4) (Figure 2). Because most patients in our cohort received blinatumomab before inotuzumab (blinatumomab mAb1/inotuzumab mAb2), we focused the analysis on 25 patients who received blinatumomab as the mAb1 and inotuzumab as the mAb2 therapy. Among these 25 patients who received blinatumomab mAb1, the 12-month EFS post-blinatumomab mAb1 was 16.0% (95% CI, 6.5-39.2), which was similar between CD19neg/dim (9.1%; 95% CI, 1.4-58.9) and CD19pos (21.4%; 95% CI, 7.9-58.4) relapse (P = .48) (Figure 3A). OS post-mAb1 relapse was comparable between CD19neg/dim and CD19pos relapse post-blinatumomab mAb1 (1-year OS, 45.5% [95% CI, 23.8-86.8] vs 35.7% [95% CI, 17.7-72.1]; P = .73) (Figure 3B). There was no difference in 1-year OS between patients who achieved CR after blinatumomab mAb1 and those who did not (38.5% [95% CI, 19.3-76.5] vs 41.7% [95% CI, 21.3-81.4]; P = .61), and between patients who received (46.2%; 95% CI, 25.7-83.0) and did not receive (33.3%; 95% CI, 15.0-74.0) interim non–mAb-based salvage therapy for relapse after blinatumomab mAb1 (P = .82) (Figure 3C). OS after post-blinatumomab mAb1 relapse was similar between patients who responded and did not respond to inotuzumab mAb2 (1-year OS, 41.2% vs 37.5%; P = .71). Survival in 4 patients who received inotuzumab mAb1 and blinatumomab mAb2 was not analyzed because of the small number of patients.
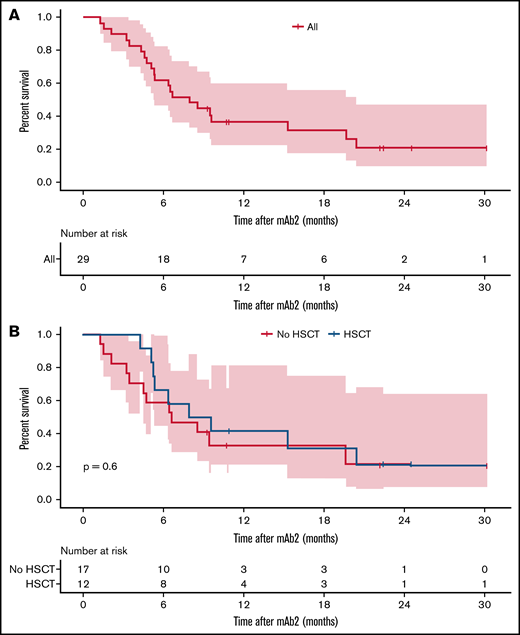
OS after mAb2 therapy. (A) All patients. (B) According to transplant status after mAb2 therapy.
OS after mAb2 therapy. (A) All patients. (B) According to transplant status after mAb2 therapy.
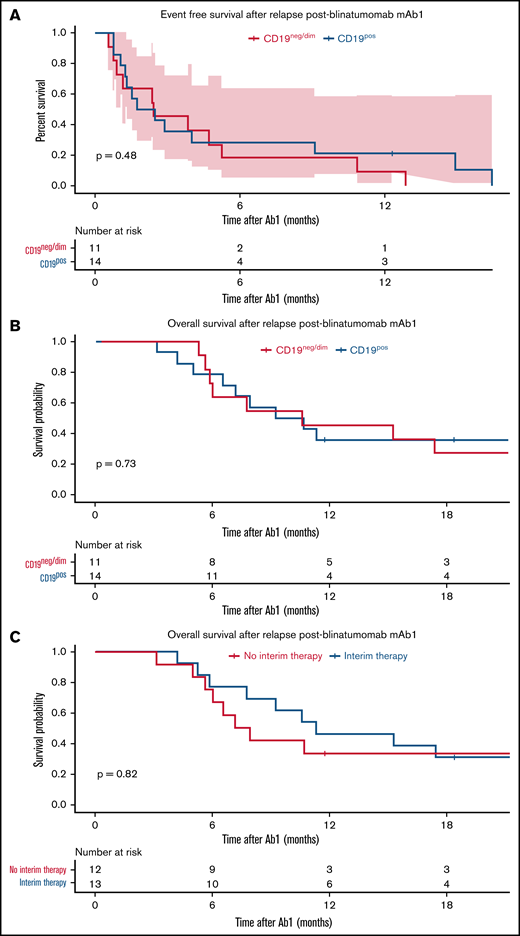
Survival after post-blinatumomab mAb1 relapse. (A) EFS as stratified according to the CD19 expression post-blinatumomab relapse. (B) OS as stratified according to the status of CD19 expression post-blinatumomab relapse. (C) OS as stratified according to receipt of interim salvage therapy between mAb1 and mAb2.
Survival after post-blinatumomab mAb1 relapse. (A) EFS as stratified according to the CD19 expression post-blinatumomab relapse. (B) OS as stratified according to the status of CD19 expression post-blinatumomab relapse. (C) OS as stratified according to receipt of interim salvage therapy between mAb1 and mAb2.
Among 12 patients who underwent alloHSCT after mAb2, 4 patients (33.3%) experienced disease relapse. At a median follow-up of 18 months, 9 patients (75.0%) have died (5 of 7 in myeloablative transplant and 4 of 5 in non-myeloablative transplant), and 3 patients remained alive at last follow-up (2 in ongoing remission and 1 with active disease). Causes of death in alloHSCT patients included disease progression (n = 4), infection (n = 1), and transplant-related toxicity (severe aGVHD, n = 2; SOS, n = 1; transplant-related organ toxicities, n = 1). The 1-year EFS and OS after alloHSCT were 25.0% and 31.2%, respectively (Figure 4). Number of treatments before alloHSCT or the receipt of other interim non-mAb salvage therapies between mAb1 and mAb2 did not affect the post-alloHSCT outcomes after mAb2.
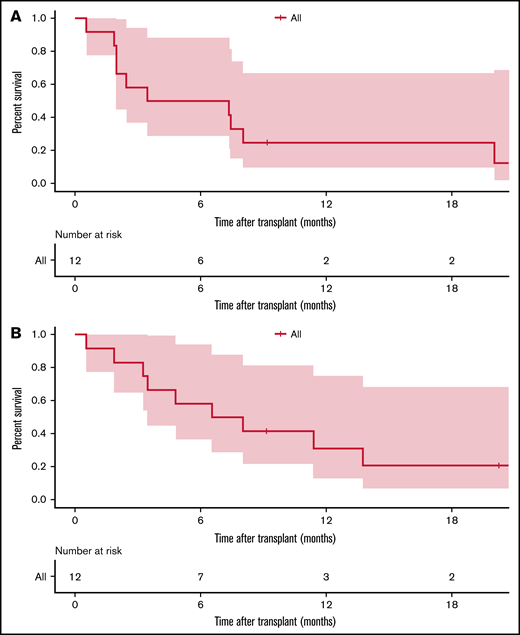
Kaplan-Meier survival estimate after alloHSCT in 12 patients who previously received blinatumomab and inotuzumab ozogamicin. (A) EFS. (B) OS.
Kaplan-Meier survival estimate after alloHSCT in 12 patients who previously received blinatumomab and inotuzumab ozogamicin. (A) EFS. (B) OS.
For 17 patients who did not undergo alloHSCT after mAb2, 12 (70.6%) died at the time of data cutoff, all due to progressive disease (n = 12). Five of 17 patients remain alive, including 1 patient in CR from salvage therapy (with inotuzumab re-treatment) and 4 patients with persistent disease (2 with systemic disease and 2 with isolated central nervous system relapse). The median OS in patients who did not undergo alloHSCT after mAb2 was 203 days (95% CI, 140-not reported) with the corresponding 1-year OS after mAb2 of 26.5% (95% CI, 11.2-62.4). Patients who achieved CR to salvage therapy after post-mAb2 relapse had a trend toward superior OS compared with patients who did not (median, 261 vs 145 days; P = .06).
Discussion
The current study describes the characteristics and outcomes of patients with R/R B-ALL who received both blinatumomab and inotuzumab for relapsed disease, including a subset of patients who had alloHSCT after both mAbs. The majority of patients in our cohort received blinatumomab as the first mAb, with response rates comparable to those published previously,14 and inotuzumab was used as the next immediate salvage therapy in 50% of the patients who relapsed after blinatumomab. We found that inotuzumab was effective in inducing CR in 68% of the patients who relapsed after blinatumomab regardless of CD19 expression status. However, despite transplant realization rate of 65% in patients who received blinatumomab mAb1 and inotuzumab mAb2, long-term survival was limited due to high transplant-related mortality (TRM) and relapse rates.
We observed a high rate of CD19neg/dim relapse (44%) after blinatumomab in our cohort. Prior studies of blinatumomab in B-ALL reported incidences of CD19neg relapses ranging from 10% to 40%,9,15 with some studies suggesting worse outcome in CD19neg relapse patients.16,17 Although data on cytogenetic abnormalities at the time of CD19neg/dim relapse were not available, we observed no evidence of mixed lymphoid lineage rearrangement in these patients at the time of diagnosis. We also observed no significant difference in outcomes between CD19neg/dim and CD19pos relapse post-blinatumomab mAb1. Both groups responded equally well to inotuzumab mAb2, and post-alloHSCT outcome was comparable.
Because most of our patients received blinatumomab before inotuzumab, we focused our analysis on these 25 patients with blinatumomab mAb1 and inotuzumab mAb2. Inotuzumab mAb2 was an effective salvage therapy for patients who relapsed after blinatumomab mAb1 irrespective of CD19 expression or receipt of other interim salvage therapy between mAb1 and mAb2. The CR rate after inotuzumab mAb2 treatment was 68%, comparable to CR rates observed with inotuzumab when used in salvage 1 and 2 and blinatumomab naive settings.18-20 Inotuzumab also yielded comparable responses whether it was used immediately following blinatumomab or after interim salvage therapy post-blinatumomab. However, the response was short-lived, and most patients who did not have alloHSCT after achieving CR following mAb2 eventually relapsed, similar to data from the INO-VATE (INotuzumab Ozogamicin trial to inVestigAte Tolerability and Efficacy) study.4 These findings indicate the high antileukemic activity of inotuzumab in these heavily treated, post-blinatumomab relapse patients regardless of CD19 expression status and support its use as an effective bridge to alloHSCT.21-24
Although several studies have reported favorable outcomes in patients treated with sequential treatments of inotuzumab-based chemotherapy combination followed by blinatumomab,25 four patients in our study received inotuzumab immediately followed by blinatumomab consolidation but all experienced disease relapse. Although limited by the extremely small number of patients, this finding raises caution against this treatment approach in the setting of multiply relapsed B-ALL and suggests more data are needed on effective consolidative therapeutic strategy.
In our cohort, ∼60% of patients who achieved CR after mAb2 proceeded to alloHSCT. The incidence of aGVHD in our cohort was high. However, due to a small number of transplant patients in our cohort and heterogeneous alloHSCT platforms, further analysis to determine risk of aGVHD was not statistically justified. In our cohort, we observed 40% TRM, higher than the rate reported in the INO-VATE and TOWER (Blinatumomab Versus Standard of Care Chemotherapy in Patients With Relapsed or Refractory Acute Lymphoblastic Leukemia) studies.21,25 However, the high TRM rate in this study reproduced the previous report from our group, which showed a poor posttransplant outcome and high TRM in patients who had alloHSCT after achieving CR3.26 Another small retrospective study looking at the outcomes of alloHSCT in adult B-ALL after blinatumomab salvage therapy reported 1-year OS and TRM of 77% and 18%, respectively.22 The high TRM and low OS of alloHSCT in our cohort are likely attributed to the biased selection of heavily treated patients, who deliberately did not pursue immediate consolidative alloHSCT after initial treatment with either blinatumomab or inotuzumab, and reflect a major challenge in the management of these difficult-to-treat patients. Due to the limited number of patients and different alloHSCT platforms, we could not analyze prognostic factors for survival after alloHSCT, including a comparison between transplant and nontransplant cohorts. However, it is notable that ∼25% of non-transplanted patients were able to attain durable leukemic control more than 6 months and remained alive at the time of last follow-up.
Because most patients in our series received inotuzumab as the mAb2 therapy, SOS was one of the relevant complications of special interest. We observed hepatic SOS in 4 patients (13.3%), with most events occurring in the post-alloHSCT setting (1 pre-alloHSCT and 3 post-alloHSCT), including one case of grade 5 SOS post-alloHSCT. The median time from inotuzumab to alloHSCT was 40 days. Three patients who developed SOS post-alloHSCT had alloHSCT at 23, 40, and 44 days after the last dose of inotuzumab. The incidence of SOS in other studies of inotuzumab and alloHSCT ranges from ∼10% to 20%.21,27,28 In the largest series of alloHSCT after inotuzumab, the incidence of SOS was 18.8% with the median onset date from the last dose of inotuzumab to the onset of SOS of 58 days.21 Our study therefore suggests that although TRM was relatively high in these patients, hepatic SOS occurred at a rate comparable to the other published series and was not the major contributing factor to TRM. This finding suggests the importance of inotuzumab dose intensity and implication of proper patient selection for alloHSCT, including interval from inotuzumab to transplant or intensity/choice of conditioning regimens to alleviate the risk of TRM.
The major strength of our report is the comprehensive description of patients with R/R B-ALL who received salvage treatment with blinatumomab followed by inotuzumab and inclusion of outcomes in patients who underwent alloHSCT afterward. The major limitations of this study were the small number of patients, patient selection bias, and missing information due to the retrospective nature of the cohort. The heterogeneity of salvage therapy, concomitant treatments, and alloHSCT platforms can confound the interpretation of the study. Data on MRD were not readily available in all patients, thus potentially affecting the significance of results. Lastly, because most patients in our study received blinatumomab as the mAb1 followed by inotuzumab as the mAb2, the response pattern and toxicity profile after mAb2 in our cohort could be more representative of an effect of inotuzumab. The recent retrospective study by Badar et al7 reported efficacy and toxicity of blinatumomab and/or inotuzumab in patients with R/R B-ALL. The study included a subset of 61 patients who received both mAbs (n = 40, blinatumomab as mAb1; n = 21, inotuzumab as mAb1), but the analysis focused mainly on response duration and survival as related to the mAb sequence administered. Due to a small number and treatment bias toward blinatumomab mAb1, we were not able to explore the impact of mAb sequence on clinical outcomes in our study. However, we focused on management and outcomes of patients who relapsed after or developed resistance to both mAbs. We provide more comprehensive information on interim therapies between the two mAbs, CD19 and CD22 expression changes, post-mAb2 treatments (especially alloHSCT), and toxicities than what was described in the study by Badar et al. The details of post-mAb2 treatments, including alloHSCT and CD19 CAR T cells, as well as subsequent clinical outcomes provided in our study are critical to help clinicians assess prognosis and guide clinical development of novel therapeutic strategies following blinatumomab and inotuzumab.
Patients with R/R B-ALL progressing after both blinatumomab and inotuzumab represent a group of ultra-high-risk patients. Inotuzumab appears to induce high CR rates in patients resistant to blinatumomab, and subsequent alloHSCT offers potential long-term remission in a subset of these patients. However, the high incidence of TRM and relapse remains significant challenges. Although alloHSCT may offer a survival benefit to a subset of these heavily treated patients, studies exploring patient selection for transplant and novel, less-toxic conditioning regimens are warranted to improve alloHSCT outcomes. Moreover, with the recent approval of a CD19 CAR T-cell therapy for adults with R/R B-ALL,29 prospective evaluation will be needed to examine the of role of consolidative CD19 CAR T-cell therapy after blinatumomab and inotuzumab, and comprehensive analysis on optimal sequence of CD19- and CD22-directed therapies will be vital in establishing new, more effective treatment paradigms for patients with R/R B-ALL.
Acknowledgments
All authors are thankful for the patients and their families for granting the consent to participate in research studies.
K.W. receives salary support from Parker Institute for cancer immunotherapy at Memorial Sloan Kettering Cancer Center, New York, and Faculty of Medicine, Chulalongkorn University, Bangkok Thailand. This study is supported in part by the National Institutes of Health, National Center for Advancing Translational Sciences award P01 CA23766, and National Cancer Institute Cancer Center Support grant P30 CA008748.
Authorship
Contribution: K.W. and J.H.P. designed the study and wrote the paper; M.R. reviewed the pathology data; K.W. collected the data and conducted the analysis; A.C.K., M.B.G., and Y.B. participated in data collection; K.W., Y.B., and J.H.P. conducted the statistical analysis; A.C.K., M.B.G., B.G., M.-A.P., and J.H.P. took care of the patients; and all the authors reviewed and approved the paper.
Conflict-of-interest disclosure: A.C.K. serves on a scientific advisory board for Astellas; and has received consulting fees from AbbVie. M.B.G. receives research support from Amgen, Actinium Pharmaceuticals, and the National Cancer Institute (R42 CA254685-01); and serves in an advisory role for Sanofi. B.G. reports research funding for a clinical trial from Actinium Pharma. M.-A.P. reports honoraria from Kite/Gilead, AbbVie, Bellicum, Bristol Myers Squibb, Incyte, Merck, Novartis, Nektar Therapeutics, Omeros, and Takeda; serves on Data and Safety Monitoring Boards for Servier and Medigene and the scientific advisory boards of MolMed and NexImmune; has received research support for clinical trials from Incyte, Kite/Gilead, and Miltenyi Biotec; and serves in a volunteer capacity as a member of the Board of Directors of the American Society for Transplantation and Cellular Therapy and Be the Match (National Marrow Donor Program), as well as on the CIBMTR Cellular Immunotherapy Data Resource Committee. J.H.P. receives funding from the Conquer Cancer Foundation of ASCO, a Leukemia and Lymphoma Society Career Development Grant, the Geoffrey Beene Cancer Foundation, a National Comprehensive Cancer Center Young Investigator Award, and an American Society of Hematology Scholar Junior Faculty Award; and has consulted and provided an advisory role for Amgen, Novartis, Kite Pharma, Incyte, Allogene, Autolus, Intellia, Artiva, AstraZeneca, Umoja, Kura Oncology, Innate Pharma, Takeda, and Servier. The remaining authors declare no competing financial interests.
Correspondence: Jae H. Park, Leukemia Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10021; e-mail: parkJ6@mskcc.org.

