

Abstract
In the 1960s, Dr Jan Waldenström argued that patients who had monoclonal proteins without any symptoms or evidence of end-organ damage represented a benign monoclonal gammopathy. In 1978, Dr Robert Kyle introduced the concept of “monoclonal gammopathy of undetermined significance” (MGUS) given that, at diagnosis, it was not possible with available methods (ie, serum protein electrophoresis to define the concentration of M-proteins and microscopy to determine the plasma cell percentage in bone marrow aspirates) to determine which patients would ultimately progress to multiple myeloma. The application of low-input whole-genome sequencing (WGS) technology has circumvented previous problems related to volume of clonal plasma cells and contamination by normal plasma cells and allowed for the interrogation of the WGS landscape of MGUS. As discussed in this chapter, the distribution of genetic events reveals striking differences and the existence of 2 biologically and clinically distinct entities of asymptomatic monoclonal gammopathies. Thus, we already have genomic tools to identify “myeloma-defining genomic events,” and consequently, it is reasonable to consider updating our preferred terminologies. When the clinical field is ready to move forward, we should be able to consolidate current terminologies—from current 7 clinical categories: low-risk MGUS, intermediate-risk MGUS, high-risk MGUS, low-risk smoldering myeloma, intermediate-risk smoldering myeloma, high-risk smoldering myeloma, and multiple myeloma—to future 3 genomic-based categories: monoclonal gammopathy, early detection of multiple myeloma (in which myeloma-defining genomic events already have been acquired), and multiple myeloma (patients who are already progressing and clinically defined cases). Ongoing investigations will continue to advance the field.
Learning Objectives
Review of current clinical risk scores for myeloma precursor conditions, which are limited indirect measures of disease burden/activity
Show how low-input WGS reveals striking differences and existence of 2 biologically and clinically distinct entities
CLINICAL CASE
In the beginning of January 2016, a 46-year-old previously healthy male lawyer was applying for a new life insurance policy. As part of the application process, the insurance company required a standard health questionnaire and laboratory blood tests. The results from the blood work showed a normal complete blood count and a normal comprehensive metabolic panel with the exception of an elevated total protein of 8.9 g/dL (normal reference range, 6.0-8.3 g/dL). The patient was referred to a hematologist, who ordered repeat laboratory blood tests, including additional serum immune assays, to further investigate the elevated total protein levels. The repeated results confirmed normal complete blood count and comprehensive metabolic panel, whereas a serum protein electrophoresis (SPEP) revealed evidence of a monoclonal band in the gamma region and immunofixation showed immunoglobulin G (IgG) κ isotype. The concentration of the monoclonal (M)–protein was determined to 0.9 g/dL. Quantitative immunoglobulins showed normal IgG, immunoglobulin A (IgA), and immunoglobulin M (IgM) levels. Also, serum free light chains (FLCs) were evaluated, and they showed normal levels. Given the low concentration of the M-protein, the IgG isotype, the absence of an abnormal FLC ratio, and normal IgA (150 mg/dL) or IgM (173 mg/dL) concentrations, per current clinical guidelines,1 the hematologist recommended no bone marrow biopsy and no imaging. Instead, the patient was recommended to repeat the bloodwork in 6 months. The patient was given the diagnosis of monoclonal gammopathy of undetermined significance (MGUS), and he was told that the lifetime risk of progression to multiple myeloma (MM) was very low.
When the patient returned for a follow-up visit in July 2016, all the results were virtually unchanged. Per current clinical guidelines,1 the hematologist now recommended follow-up annually with repeated bloodwork. When the patient returned in July 2017, the IgG κ M-protein had increased to 1.1 g/dL, whereas all other results were similar to the year before. The patient had no symptoms. In July 2018, he returned for a follow-up visit, and the bloodwork now revealed an IgG κ M-protein of 1.4 g/dL. The FLC κ levels were slightly elevated at 5.3 mg/dL (normal reference range, 0.33-1.94 mg/dL), FLC λ was 0.8 mg/dL (normal reference range, 0.57-2.63 mg/dL), and the FLC ratio was 6.63 (normal reference range, 0.26-1.65). Also, the IgM concentration was decreased at 25 mg/dL (normal reference range, 37-286 mg/dL), whereas IgA was within the normal reference interval at 78 mg/dL (61-356 mg/dL). Given the gradual worsening of serum immune markers, the hematologist discussed with the patient that he may choose to undergo a bone marrow biopsy and imaging to rule out MM, but the patient asked his doctor if it was totally necessary since he felt completely healthy, ate healthy food, and exercised several days every week. After a longer discussion, the patient and the hematologist decided to hold off with additional testing and continue with annual laboratory results.
In June 2019, about 3.5 years after initial diagnosis of MGUS, the patient came for a visit to review his annual bloodwork. Now the SPEP showed an IgG κ M-protein of 1.6 g/dL. The FLC κ levels were again elevated now at 8.6 mg/dL (normal reference range, 0.33-1.94 mg/dL), FLC λ was 0.5 mg/dL (normal reference range, 0.57-2.63 mg/dL), and the FLC ratio was 17.2 (normal reference range, 0.26-1.65). The IgM concentration was further decreased at 18 mg/dL (normal reference range, 37-286 mg/dL), and IgA was decreased at 40 mg/dL (61-356 mg/dL). Hemoglobin was slightly decreased at 13.2 g/dL (normal reference range, 13.5-17.5 g/dL), whereas all other laboratory results (including calcium, creatinine, albumin, lactate dehydrogenase, and β2- microglobulin) were normal. The hematologist recommended the patient to undergo a bone marrow biopsy and a positron emission tomography/computed tomography to rule out MM. Immunohistochemistry staining of the core biopsy specimen showed 30% κ light chain–restricted plasma cells in the bone marrow. Also, whole-body positron emission tomography/computed tomography revealed a 1.1-cm diameter lytic lesion in ileum on the right side of the pelvis with an standardized uptake value (SUV) of 6.7, a 1.2-cm diameter lytic lesion in the left femur with a SUV of 8.2, and 2 small (<5 mm diameter) lytic lesions in the left fifth and sixth ribs with SUVs of 2.5 and 3.1, respectively. Fluorescence in situ hybridization and single-nucleotide polymorphism array testing of the bone marrow aspirate did not capture any high-risk characteristics. Ten-color flow cytometry of the bone marrow aspirate confirmed the immunophenotype of κ light chain–restricted plasma cells expressing CD56 and CD117 but negative for CD20. The patient was diagnosed with standard-risk MM and started combination therapy 2 weeks later, after he had undergone routine workup with a clinical exam and baseline echocardiogram and electrocardiogram.
Because most MGUS cases will never progress, statistically speaking, this patient case illustrates a relatively uncommon situation with progression to MM within a few years from initial MGUS diagnosis. However, most physicians who monitor large numbers of patients with MGUS have seen these kinds of cases in their clinic. This is reflective of the following 2 facts: (1) most cases with MGUS remain stable over time, but at the same time, (2) all MM cases are consistently preceded by MGUS (but the majority do not know they had MGUS prior to MM because no testing was done).2 When it comes to current clinical risk scores to predict progression from MGUS to MM, it is important to emphasize that today's risk scores only provide the average risk of progression for all individuals with a given score. None of the available risk scores provide the absolute risk of progression for an individual patient. Furthermore, current clinical risk scores are based on tumor burden, and they are unable to detect progressors among MGUS cases with lower disease burden. Indeed, current clinical risk scores show that individuals with higher disease burden (higher plasma cell percentage and/or higher serum immune marker concentrations) on average have a higher probability of progressing compared with the average individual with lower disease burden. Although lower disease burden has lower risk of progression, just like the above patient case, there are individuals with lower disease burden progressing to MM. Conversely, many individuals with higher disease burden will never progress.
There is an unmet clinical need for better assays allowing the physician to determine the individual patient's risk of progression to MM. We do not yet have any such established assays available in the clinic. The best tools we have today include longitudinal monitoring and reassessments, as discussed in detail in this chapter. This review addresses current status of science and clinical management, novel and emerging technologies, recent discoveries, and future directions.
Initial observations and emergence of different schools of thought
Early discovery work focusing on abnormal serum proteins was pioneered by Dr Jan Waldenström and colleagues. In 1944, he presented his paper “Incipient Myelomatosis or ‘Essential’ Hyperglobulinemia With Fibrinogenopenia—A New Syndrome?” in which he described 3 patients with refractory anemia and bleeding tendency whose sera exhibited a very high viscosity. Waldenström speculated that a special globulin fraction was the cause of the increased viscosity, and by ultracentrifugation studies, he was able to demonstrate that the sera of 2 of these patients contained macroglobulins (ie, plasma globulins of high molecular weight)—subsequently referred to as “Waldenström's macroglobulinemia.”3 Together with Dr Carl-Bertil Laurell—a world-class clinical chemist—Waldenström started exploring conditions with gammaglobulin derangements in a systematic manner, as well as clinical correlates of monoclonal and polyclonal gammopathies.4 Through their translational research efforts, they delineated the occurrence and clinical significance of so-called monoclonal components. Gradually, observations from the laboratory coupled with clinical data led to the emergence of 2 major schools of thought. In the 1960s, Waldenström proposed that there were patients who had monoclonal (M)–proteins without any symptoms or evidence of end-organ damage, representing a benign monoclonal gammopathy (MG).5-8 Waldenström was of the firm belief that benign MG was unrelated to MM. Conversely, the alternate opinion was that some patients with asymptomatic monoclonal proteins nevertheless progressed over time to MM and that it was important to not term the process entirely benign. In 1978, Dr Robert Kyle published his observations from a retrospective chart review of all individuals (N = 241) diagnosed with a MG at the Mayo Clinic prior to January 1, 1971. In brief, among the 241 cases, he found that after a 5-year follow-up period, (1) the M-protein remained stable in 137 (57%) patients; (2) the M-protein increased by 50% or more in 22 (9%) patients; (3) onset of MM, Waldenström macroglobulinemia, or amyloid light chain amyloidosis occurred in 27 (11%) patients; and (4) 55 (23%) patients died without 5-year follow-up serum.6 When he compared the mean M-protein concentration and percent bone marrow plasma cell infiltration at baseline for the 4 groups, there were no differences (mean concentrations were 1.6-1.8 g/dL and mean plasma cell infiltration in bone marrow aspirates was 3%-4% across the 4 groups).6 Based on these small numbers, Kyle6 observed that among individuals with an M-protein and who later developed MM, the size of the monoclonal peak increased along with symptoms prior to onset of MM. Therefore, Kyle argued in his seminal paper from 1978 that “monoclonal gammopathy of undetermined significance” (MGUS) is a better term because one cannot tell whether the monoclonal protein will remain unchanged or whether the patient has an evolving MM. The word undetermined was used to reflect that, at diagnosis, it was not possible with available methods (ie, SPEP to define the concentration of M-proteins and microscopy to determine the plasma cell percentage in bone marrow aspirates) to determine which patients would ultimately progress to MM.
Diagnostic criteria and types of abnormal serum proteins
The current definition of MGUS is characterized by the presence of M-proteins or an abnormal FLC ratio in peripheral blood.1 For an individual to be diagnosed with MGUS, per current definitions, the concentration of the monoclonal spike (M-spike) has to be less than 3 g/dL, and for an individual to be diagnosed with light chain MGUS, the FLC ratio has to be abnormal (normal reference for κ/λ FLC ratio, 0.26-1.65), but the involved/uninvolved ratio has to be less than 100.9 It should be emphasized that elevated FLC concentrations are not unique to plasma cell disorders, and a clinical interpretation of the results is always required. For example, elevated FLC levels can be reflective of underlying renal insufficiency, autoimmune conditions, systemic inflammation, infection, and other causes. Furthermore, if the individual undergoes a bone marrow biopsy, based on current diagnostic criteria, the plasma cell involvement of the bone marrow must be less than 10%.1 Last, the clinical workup must be negative for evidence of end-organ damage from plasma cell dyscrasia, hypercalcemia, anemia, renal failure, lytic bone lesions, or multiple (2 or more) focal lesions in the skeleton by magnetic resonance imaging.9 If 1 or more of those abnormalities are identified, unless there is another explanation for the abnormality (eg, anemia due to bleeding, renal failure due to cardiovascular disease), then the patient would be diagnosed with MM. If none of the above-listed abnormalities are present, but the M-spike is 3 g/dL or more and/or there is over 10% (but less than 60%) plasma cells in the bone marrow; then, based on current criteria, the diagnosis would be smoldering myeloma.1,9
Using a combination of serum-based protein assays (SPEP, immunofixation, and serum FLC assays), MGUS cases can be classified based on the isotype of M-proteins present. So-called non-IgM MGUS is the most common type and is defined by the presence of IgG, IgA, and, rarely, immunoglobulin D or immunoglobulin E M-proteins.10 IgM MGUS is defined by the presence of IgM M-proteins.11 Light chain MGUS is defined by an abnormal FLC ratio, indicating an excess of monoclonal FLCs in the absence of M-proteins.12 Non-IgM MGUS and light chain MGUS are caused by monoclonal bone marrow plasma cells, and they are precursors of MM.2 IgM MGUS is commonly caused by monoclonal lymphoplasmacytic cells and is a precursor to other lymphoproliferative disorders, most notably Waldenström macroglobulinemia, chronic lymphocytic leukemia, and, only in very rare instances (<0.5%), MM.11 In addition, MGUS of all types, especially λ light chain MGUS, can precede amyloid light chain amyloidosis.13 It should be noted that in the general MM population, around 80% of patients have an M-protein and approximately 20% have light chain secretory MM (ie, without evidence of an M-protein).14 Among patients with MM who have an M-protein, most of them also have an abnormal FLC ratio caused by overproduction of either κ or λ FLCs.14 Among patients diagnosed with MGUS with an M-spike, around 30% also have an abnormal FLC ratio.15
Epidemiologic studies: novel insights
In retrospective studies with long-term follow-up, Kyle et al10 and others16 have reported 0.5% to 1.0% annual average risk of progression from MGUS to MM. Importantly, most retrospective studies seeking to identify risk factors for progression are based on statistical models using risk factor data from a single time point (usually the initial workup).10,16 Data from this kind of modeling have been used to develop clinical consensus guidelines recommending annual peripheral blood monitoring of serum protein markers and other assays for patients with intermediate-risk and high-risk MGUS.1 Inspired by the observations by Kyle in 1978,6 a few smaller retrospective studies17,18 have proposed evolving changes in M-protein levels are associated with progression to MM. However, in clinical practice, most patients are typically counseled based on their risk profile captured at initial workup. Regarding cases with light chain MGUS, there is only limited information available regarding the risk of progression from light chain MGUS to light chain MM, and consequently, clinical guidelines for this condition are lacking.12 However, the biggest limitation of defining risk in this fashion is lead-time bias because there are no primary care screening guidelines. Therefore, patients are often found to have MGUS after an incidental routine laboratory finding (eg, as part of the workup for elevated total protein). Alternatively, patients may be diagnosed with MGUS after a workup for non-myeloma-defining signs/symptoms (eg, unexplained peripheral neuropathy or slight increase in serum creatinine).
In 2009, the first prospective population-based MGUS screening study was published. Using stored peripheral blood samples that were collected as part of the large (N = 77 469) National Cancer Institute Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (NCI PLCO), this large study was able to show that MM is consistently preceded by the MGUS precursor stage.2 This important observation definitively links MM to its precursor disease. This study has set the stage for future investigations seeking to define molecular mechanisms of progression as well as efforts designed to develop clinical biomarkers.
In 2019, a longitudinal follow-up study, also based on the NCI PLCO trial, investigated dynamics of serum immune markers (including monoclonal proteins, FLCs, and quantitative immunoglobulins) in individuals with MGUS.19 The follow-up study provided novel insights by illustrating that an individual patient's risk of progression from MGUS to MM is not constant. For individuals with light chain MGUS, the same patterns were observed. The 2019 study was based on prospectively collected annual samples and showed that individual patients' clinical risk categories could convert from lower to a higher risk over time. Indeed, the data show how low-risk or intermediate-risk MGUS cases can convert into high-risk MGUS and progress to MM within 5 years. For individuals with light chain MGUS, the same conversion patterns were observed. These findings are clinically important and support the application of annual follow-up visits with blood testing and reassessment of a patient's clinical risk status as derived from serum immune markers in peripheral blood. This is true for all individuals diagnosed with MGUS or light chain MGUS.
In the longitudinal follow-up NCI PLCO study,19 first, a cross-sectional marker analysis was conducted. In the statistical model, serum immune markers proposed in previous studies were included.10,16 The risk of progression in association with each marker was analyzed using the prediagnostic measurements from the time point most proximal to the MM diagnosis date. Second, a longitudinal analysis was carried out to define patterns of serum marker changes. A scoring system was developed based on accumulated points by using the results from the cross-sectional analyses. For MGUS, the following variables were defined as risk factors for progression (Table 1): M-spike concentration of 1.5 g/dL or more (1 point), M-spike with IgA isotype (1 point), serum FLC ratio less than 0.1 or more than 10 (1 point), and immunoparesis (≥1 uninvolved immunoglobulin below lower limit of normal; 1-2 points). For the first time, a scoring system was developed for light chain MGUS. For light chain MGUS, the following variables were defined as risk factors for progression: serum FLC ratio less than 0.1 or more than 10 (1 point) and immunoparesis (1, 2, or 3 points) (Table 1). The score was defined as the total (accumulated) number of points assigned to risk factors for each individual blood sample. The scores were defined as follows (Table 1): MGUS (0-1, low-risk and low-risk light chain; 2, intermediate-risk and intermediate-risk light chain; ≥3, high-risk and high-risk light chain). Interestingly, when using the score to define risk of progression, 53% of patients with progressing MGUS but only 1 of 108 patients with nonprogressing MGUS had a high-risk score (Figure 1).9 Similarly, high-risk status was found in 70% of patients with progressing light chain MGUS, but only 1 of 120 patients with nonprogressing light chain MGUS had a high-risk score (Figure 2). Clinically important, the study found that most individuals who developed MM after a preceding state of high-risk MGUS had converted from low-risk or intermediate-risk stages within 5 years before MM diagnosis. For individuals with light chain MGUS progression, the patterns were very similar. Although most individuals who progressed from MGUS or light chain MGUS to MM or light chain MM, respectively, were characterized by rising serum immune markers within a 5-year time window, a small fraction of cases had rising serum immune markers for less than 1 year. In fact, 4 of 43 (9%) individuals who progressed from MGUS to MM had a low-risk score 1 year prior to MM diagnosis. Similarly, 1 of 10 (10%) patients with progressing light chain MGUS were low risk 1 year prior to light chain MM diagnosis (Figures 1 and 2). Furthermore, the study found that only 21% of individuals who progressed from MGUS to MM fulfilled the blood-based criteria (ie, 3 g/dL or more M-protein) for smoldering myeloma1 prior to diagnosis of MM. Because annual bone marrow biopsies are not supported by clinical guidelines, this is clinically important information for physicians who monitor individuals diagnosed with MGUS given that most patients with MGUS who progress to MM skip the smoldering myeloma phase based on blood-based criteria.
Adverse markers for progression19
| Variable* | Adverse marker | Points |
|---|---|---|
| MGUS | ||
| M-spike isotype | IgA | 1 |
| M-spike concentration | ≥15 g/L | 1 |
| Serum FLC ratio (κ:λ) | <0.1 or >10 | 1 |
| Immunoparesis | Uninvolved immunoglobulins below lower level of normal | 1 or 2 |
| Light chain MGUS | ||
| Serum FLC ratio (κ:λ) | <0.1 or >10 | 1 |
| Immunoparesis | Uninvolved immunoglobulins below lower level of normal | 1, 2, or 3 |
| Variable* | Adverse marker | Points |
|---|---|---|
| MGUS | ||
| M-spike isotype | IgA | 1 |
| M-spike concentration | ≥15 g/L | 1 |
| Serum FLC ratio (κ:λ) | <0.1 or >10 | 1 |
| Immunoparesis | Uninvolved immunoglobulins below lower level of normal | 1 or 2 |
| Light chain MGUS | ||
| Serum FLC ratio (κ:λ) | <0.1 or >10 | 1 |
| Immunoparesis | Uninvolved immunoglobulins below lower level of normal | 1, 2, or 3 |
Risk categories for progression to MM and light chain MM: 0 to 1, low-risk and low-risk light chain; 2, intermediate-risk and intermediate-risk light chain; 3 or higher, high-risk and high-risk light chain.

Longitudinal analysis of risk scores among selected individuals with and without progression from MGUS to MM.19 Using data from the cross-sectional analysis, a risk score was developed and implemented to assess and label individual blood samples as follows: low-risk MGUS, 0 to 1 (gray); intermediate-risk MGUS, 2 (white); high-risk MGUS, 3 or higher (orange). Each series of boxes represents a unique patient, each box represents a unique blood sample, and the x-axis represents number of years before MM diagnosis (for case patients) and number of years before selection (for controls). Each box includes a number that represents the risk score for that given sample. MG indicates nonprogressing MGUS, and MM indicates cases that progressed from MGUS to MM. aFulfilled the blood-based criteria for smoldering myeloma (ie, M-spike concentration ≥3 g/dL). bFulfilled the blood-based criteria for MM (ie, FLC ratio ≥100 and involved FLC concentration ≥10 mg/dL). Reprinted by permission, Landgren et al, JAMA Oncol, 2019.19
Longitudinal analysis of risk scores among selected individuals with and without progression from MGUS to MM.19 Using data from the cross-sectional analysis, a risk score was developed and implemented to assess and label individual blood samples as follows: low-risk MGUS, 0 to 1 (gray); intermediate-risk MGUS, 2 (white); high-risk MGUS, 3 or higher (orange). Each series of boxes represents a unique patient, each box represents a unique blood sample, and the x-axis represents number of years before MM diagnosis (for case patients) and number of years before selection (for controls). Each box includes a number that represents the risk score for that given sample. MG indicates nonprogressing MGUS, and MM indicates cases that progressed from MGUS to MM. aFulfilled the blood-based criteria for smoldering myeloma (ie, M-spike concentration ≥3 g/dL). bFulfilled the blood-based criteria for MM (ie, FLC ratio ≥100 and involved FLC concentration ≥10 mg/dL). Reprinted by permission, Landgren et al, JAMA Oncol, 2019.19

Longitudinal analysis of risk among selected individuals with and without progression from light chain MGUS to light chain MM.19 A risk score was developed to evaluate and label individual blood samples as follows: low-risk light chain MGUS, 0 to 1 (gray); intermediate-risk light chain MGUS, 2 (white); high-risk light chain MGUS, 3 or higher (orange). Each series of boxes represents a unique patient, each box represents a unique blood sample, and the x-axis represents number of years before MM diagnosis (for case patients) and number of years before selection (for controls). Each box includes a number that represents the risk score. LMG indicates nonprogressing light chain MGUS, and LMM indicates progressing from light chain MGUS to light chain MM. aFulfilled the blood-based criteria for MM (ie, FLC ratio ≥100 and involved FLC concentration ≥10 mg/dL). Reprinted by permission, Landgren et al, JAMA Oncol, 2019.19
Longitudinal analysis of risk among selected individuals with and without progression from light chain MGUS to light chain MM.19 A risk score was developed to evaluate and label individual blood samples as follows: low-risk light chain MGUS, 0 to 1 (gray); intermediate-risk light chain MGUS, 2 (white); high-risk light chain MGUS, 3 or higher (orange). Each series of boxes represents a unique patient, each box represents a unique blood sample, and the x-axis represents number of years before MM diagnosis (for case patients) and number of years before selection (for controls). Each box includes a number that represents the risk score. LMG indicates nonprogressing light chain MGUS, and LMM indicates progressing from light chain MGUS to light chain MM. aFulfilled the blood-based criteria for MM (ie, FLC ratio ≥100 and involved FLC concentration ≥10 mg/dL). Reprinted by permission, Landgren et al, JAMA Oncol, 2019.19
Clinical management and clinical studies
Current consensus guidelines recommend indefinite follow-up of individuals diagnosed with MGUS or smoldering myeloma.1 Longitudinal, repeated monitoring with blood markers is supported by the abovementioned prospective study that was designed to investigate dynamics of serum immune markers in individuals with MGUS.19 Also, 3 recent observational studies from Sweden and the United States have consistently demonstrated that individuals with known MGUS prior to the diagnosis of MM have about 15% better overall survival in MM (compared with individuals diagnosed with MM without knowledge of a prior MGUS diagnosis).20-22 These observations indicate that clinical follow-up of precursor disease results in earlier detection and diagnosis of MM, resulting in fewer patients with symptomatic end-organ damage and, in turn, decreased morbidity at the time of MM diagnosis, which likely contributed to the observed improvement in overall survival. In 2014, the definition of MM was expanded to include myeloma-defining biomarkers in asymptomatic individuals most likely to develop symptomatic myeloma in 2 years.9 With the advent of newer, more effective, and less toxic drugs, overall survival has improved significantly in MM.23 Three randomized controlled trials starting therapy at the stage of smoldering myeloma have shown improved progression-free survival, and 1 study showed improved overall survival.24-26 Currently, only about 5% of all patients diagnosed with MM have a previously identified precursor disease (ie, MGUS or smoldering myeloma), which limits the implementation of early treatment in most patients.20,22 To address the value of screening for MGUS in the general population, a large population-based randomized screening (monitoring vs no monitoring) study is ongoing in Iceland.27
New technologies: new opportunities
As described above in detail, clinical risk scores for myeloma precursor conditions have been developed based on indirect measurements of disease burden/activity/aggression, including bone marrow plasma cell percentage and the quantity of serum monoclonal protein.9,28-31 Although such prognostic models have proven their utility, unfortunately, they have not been useful for identifying patients with MGUS and low-risk smoldering myeloma and intermediate-risk smoldering myeloma who may have already undergone malignant transformation while the surrogate markers lag behind.9,29-31 When critically reviewing the literature, it is clear that the differentiation between MGUS and smoldering myeloma is based on the arbitrary laboratory cutoffs (above vs below 3 g/dL M-protein in peripheral blood and above vs below 10% infiltration of bone marrow plasma cells in the bone marrow) derived from retrospective medical chart review conducted 50 years ago.6,7 At the same time, based on current clinical knowledge, it is well known that some patients with MGUS can progress rapidly despite their apparent low disease burden, and conversely, many patients with smoldering myeloma will remain stable despite a higher disease burden with a behavior pattern typical of MGUS.19,28,32,33 Thus, an ability to recognize these 2 distinct clinical patterns independent of the bone marrow plasma cell percentage would offer significant advantages in clinical practice.
A range of technologies has been applied to better understand what differentiates progressive and stable myeloma precursor conditions.29,30 The application of fluorescence in situ hybridization, single-nucleotide polymorphism array, and gene expression technologies illustrates the fact that groups of patients with MGUS and smoldering myeloma with the presence of certain genomic aberrations (eg, del17p13, t(4;14)) and expression signatures have a shorter time to MM progression.34-38 The advent of next-generation sequencing has dramatically changed this scenario, allowing more comprehensive genomic investigations of individual patients, and—clinically important—providing reproducible alternatives to older tumor burden-based models. Several studies have highlighted the importance of the value of genomic events for predicting progression of the myeloma precursor conditions. For example, prior studies have identified the value of mutations in the mitogen-activated protein kinase pathway and translocations in MYC in predicting progression.29,35,37,39-42 However, until recently, technical limitations (ie, low number of clonal bone marrow plasma cells, limiting the ability to conduct sequencing assays) have forced most of these studies to only include smoldering myeloma cases and not MGUS. Although there is only limited information available, it has also been proposed that the host's biologic features and immune substrate play a role in the regulation and stability of a plasma cell neoplasm.43,44
In 2021, developments in multiparametric bone marrow plasma cell flow-sorting and the application of low-input whole-genome sequencing (WGS) technology45,46 circumvented previous problems related to volume of clonal plasma cells and contamination by normal plasma cells and allowed for the interrogation of the WGS landscape of MGUS. In the first study, 18 MGUS cases were compared with 14 smoldering myeloma and 80 MM cases.47 Given the ability of WGS to characterize single-nucleotide variants, mutational signatures, copy number variants, structural variants (SVs), and mutational signatures, differences in the genomic landscape and in the temporal acquisition of genomic events between clinically stable and progressive cases of MGUS and smoldering myeloma were characterized. Specifically, cases with a nonprogressing, clinically stable myeloma precursor condition are characterized by later initiation of the first clonal copy number changes in the patient's life and by the absence of myeloma-defining genomic events, including chromothripsis, templated insertions, mutations in driver genes, aneuploidy, and canonical apolipoprotein B mRNA-editing enzyme, catalytic polypeptide (APOBEC) mutational activity (Table 2; Figures 3 and 4).47 The distribution of genetic events revealed striking differences and the existence of 2 biologically and clinically distinct entities of asymptomatic monoclonal gammopathies: (1) 1 entity characterized by a sufficient number of myeloma genomic events to confer malignant potential and that is associated with progressive disease and (2) another entity with a lower burden of genetic events characterized by a high likelihood of a prolonged, indolent, and clinically stable course. Despite its limited sample size, this first comprehensive genomic characterization study in early myeloma disease provides proof of principle that WGS has the potential to accurately differentiate stable and progressive precursor conditions in low disease burden clinical states (Table 2; Figures 3 and 4).47 A large prospective study to confirm and expand these results has been launched.
| Myeloma-defining genomic events | Clinical entity | ||
|---|---|---|---|
| MG* | EMM† | MM | |
| Complex SV events | ✓✓ | ✓✓ | |
| Mutations in driver genes | ✓ | ✓✓ | |
| Copy number changes (ie, deletions) | ✓✓ | ✓✓ | |
| APOBEC‡ | ✓✓ | ✓✓ | |
| Early age of initiation in patient's life | ✓✓ | ✓✓ | |
| Canonical IGH translocations | ✓ | ✓✓ | ✓✓ |
| Hyperdiploidy | ✓ | ✓✓ | ✓✓ |
| MYC§ translocation | ✓ | ✓✓ | |
| Myeloma-defining genomic events | Clinical entity | ||
|---|---|---|---|
| MG* | EMM† | MM | |
| Complex SV events | ✓✓ | ✓✓ | |
| Mutations in driver genes | ✓ | ✓✓ | |
| Copy number changes (ie, deletions) | ✓✓ | ✓✓ | |
| APOBEC‡ | ✓✓ | ✓✓ | |
| Early age of initiation in patient's life | ✓✓ | ✓✓ | |
| Canonical IGH translocations | ✓ | ✓✓ | ✓✓ |
| Hyperdiploidy | ✓ | ✓✓ | ✓✓ |
| MYC§ translocation | ✓ | ✓✓ | |
Monoclonal gammopathy (MG) represents cases without evidence of genomic drivers. In the 1960s, Dr Jan Waldenström argued that patients who had monoclonal proteins without any symptoms or evidence of end-organ damage represented a benign MG.5-8 Based on careful chart reviews of individuals with monoclonal proteins, in 1978, Dr Robert Kyle introduced the concept of “monoclonal gammopathy of undetermined significance” (MGUS) given that, at diagnosis, it was not possible with available methods (ie, SPEP to define the concentration of M-proteins and microscopy to determine the plasma cell percentage in bone marrow aspirates) to determine which patients would progress to multiple myeloma (MM).6 The application of low-input whole-WGS technology45,46 has circumvented previous problems related to volume of clonal plasma cells and contamination by normal plasma cells and allowed for the interrogation of the WGS landscape of MGUS.47 As illustrated in this table, the distribution of genetic events reveals striking differences and the existence of 2 biologically and clinically distinct entities of asymptomatic monoclonal gammopathies: (1) one entity characterized by a sufficient number of myeloma genomic events to confer malignant potential and that is associated with progressive disease (early detection of multiple myeloma; EMM) and (2) another entity with a lower burden of genetic events characterized by a high likelihood of a prolonged, indolent, and clinically stable course (MG). Future prospective studies are needed to definitively address whether a (small?) proportion of cases with genomically defined47 MG eventually can convert to EMM or not. If there is only a very low rate of conversion, then the term benign monoclonal gammopathy5-8 is probably accurate for this genomically defined47 clinical entity of MG.
Early detection of multiple myeloma (EMM) are cases with MG in which genomic drivers already have been acquired.
APOBEC, as the name suggests, is a class of enzymes that was originally identified as an enzyme that edits messenger RNA species by deaminating cytosine to uracil, which in this case produces a stop codon and truncated protein. MYC includes a family of regulator genes and protooncogenes that code for transcription factors.
Activation of MYC leads to increased expression of many genes, some of which are involved in cell proliferation.
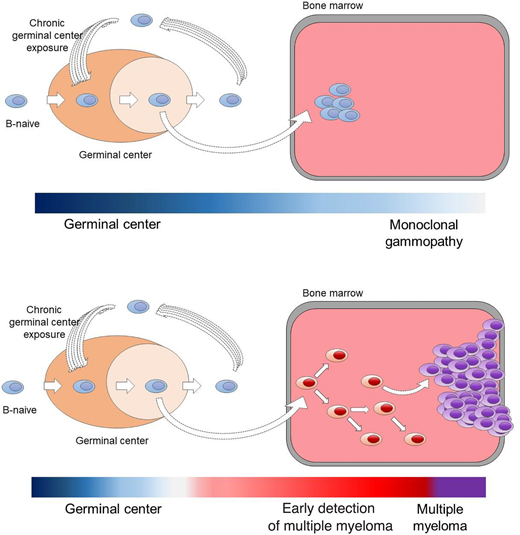
Pathogenetic models for the 2 clinically and biologically different myeloma precursor conditions (monoclonal gammopathy and early detection of multiple myeloma).29,30,47
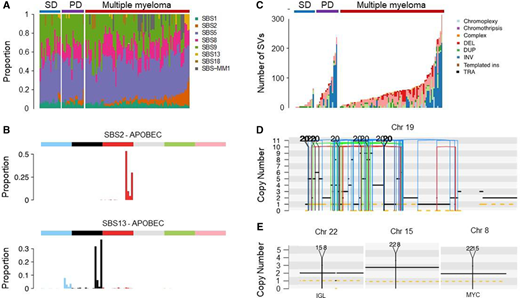
Myeloma-defining genomic events detectable only using WGS.47 (A) Mutational signatures landscape across different clinical stages. PD, progressive myeloma precursor condition (ie, early detection of multiple myeloma; EMM); SD, stable myeloma precursor condition (ie, monoclonal gammopathy; MG). (B) The 96-class profile of SBS 2 and 13 (APOBEC). (C) Prevalence of complex and single SV in MM and precursor conditions. (D) Example of chromothripsis on chromosome 19. (E) Example of templated insertion involving IGL and MYC. DEL, deletion; DUP, duplication; ins, insertions; INV, inversion; SBS, single-base substitution; TRA, translocation.
Myeloma-defining genomic events detectable only using WGS.47 (A) Mutational signatures landscape across different clinical stages. PD, progressive myeloma precursor condition (ie, early detection of multiple myeloma; EMM); SD, stable myeloma precursor condition (ie, monoclonal gammopathy; MG). (B) The 96-class profile of SBS 2 and 13 (APOBEC). (C) Prevalence of complex and single SV in MM and precursor conditions. (D) Example of chromothripsis on chromosome 19. (E) Example of templated insertion involving IGL and MYC. DEL, deletion; DUP, duplication; ins, insertions; INV, inversion; SBS, single-base substitution; TRA, translocation.
Future directions
Over 4 decades ago, Kyle6 argued that “monoclonal gammopathy of undetermined significance” (MGUS) is a preferred terminology, because one cannot tell whether the monoclonal protein will remain unchanged or whether the patient has an evolving MM. He used the word undetermined to illustrate the fact that, at diagnosis, available methods (ie, SPEP to define the concentration of M-proteins and microscopy to deter- mine the plasma cell percentage in bone marrow aspirates) were unable to determine which patients would ultimately progress to MM.6 Based on the above-described novel study showing that low-input WGS reveals progressive vs stable myeloma precursor conditions (MGUS and smoldering myeloma) as 2 distinct entities,47 it seems logical to conjecture that modern technologies in the clinic have the potential to significantly alter the management of individual patients in the near future. Development of blood-based methods is ongoing with the aim to make the identification of myeloma-defining genomic events easier in the clinical setting. Currently, there are no established validated methods, and based on limited data, the false-negative rates are too high for clinical use. More work is needed to facilitate the development of blood-based assays for identification and longitudinal tracking of genomic abnormalities. Going forward, improved and biology-oriented strategies to accurately identify patients with progressive myeloma precursor condition before clonal expansion (1) will allow earlier initiation of therapy before onset of end-organ damage or other myeloma-defining biomarkers (as defined in 20149 ) to avoid severe clinical complications and (2) will prevent patients with precursor conditions from being oversurveilled and overtreated.29,30
From a scientific perspective, my conclusion is that we already have genomic tools to identify “myeloma-defining genomic events,”47 and consequently, it is time to consider updating our preferred terminologies. When the clinical field is ready to move forward, we should be able to consolidate current terminologies—from current 7 clinical categories: low-risk MGUS, intermediate-risk MGUS, high-risk MGUS, low-risk smoldering myeloma, intermediate-risk smoldering myeloma, high-risk smoldering myeloma, and multiple myeloma—to future 3 genomic-based categories: monoclonal gammopathy, early multiple myeloma (in which myeloma-defining genomic events already have been acquired), and multiple myeloma (patients who are already progressing and clinically defined cases) (Table 2; Figures 3 and 4). Ongoing investigations will facilitate the advancement of the field with the aim to improve patient outcomes.29,30
Acknowledgments
The author thanks Sylvester Comprehensive Cancer Center Core Grant (P30 CA240139) for grant support of this work. He also thanks Paula and Rodger Riney Foundation for generous support of his research program.
Conflict-of-interest disclosure
Ola Landgren has received grant support from National Cancer Institute/National Institutes of Health, Food and Drug Administration, Leukemia and Lymphoma Society (LLS), Rising Tide Foundation, Memorial Sloan Kettering Cancer Center, Multiple Myeloma Research Foundation (MMRF), International Myeloma Foundation (IMF), Paula and Rodger Riney Foundation, Perelman Family Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, and Karyopharm; has received honoraria for scientific talks/participated in advisory boards for Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer; and served on Independent Data Monitoring Committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
Off-label drug use
Ola Landgren: nothing to disclose.
