

Abstract
The adage for smoldering myeloma (SMM) has been to observe without treatment, until criteria for active multiple myeloma were satisfied. Definitions and risk stratification models have become more sophisticated, with prognostication tailored to include high-risk cytogenetics as per the most recent International Myeloma Working Group 2020 risk model. Moreover, progress in defining genomic evolution and changes in the bone marrow microenvironment through the monoclonal continuum have given insight into the complexities underlying the different patterns of progression observed in SMM. Given recent data showing improved progression-free survival with early intervention in high-risk SMM, the current dilemma is focused on how these patients should be treated. This case-based article maps the significant advancements made in the diagnosis and risk stratification of SMM. Data from landmark clinical trials will also be discussed, and ongoing trials are summarized. Ultimately, we outline our approach to SMM and hope to impart to the reader a sound concept of the current clinical management of SMM.
Learning Objectives
Understand the risk stratification for SMM
Understand rationale for treatment vs observations and outline the current treatment options for SMM
CLINICAL CASE
A 55-year-old man presents for routine evaluation and found to have elevated total protein on serum chemistry. He had no anemia, renal impairment, or hypercalcemia. A serum protein electrophoresis and immunofixation show an IgG κ monoclonal protein measuring 2.8 g/dL, with a κ free light chain (FLC) of 40 mg/dL (FLC ratio 17). A computed tomography (CT) skeletal survey shows no lytic lesions, and no osseous lesions are found on whole-body magnetic resonance imaging (MRI). Bone marrow is normocellular, with 25% plasma cell (PC) involvement. What is his diagnosis, and how would you risk-stratify this patient?
Smoldering myeloma
Smoldering myeloma (SMM) is the asymptomatic intermediary between monoclonal gammopathy of undetermined significance (MGUS) and multiple myeloma (MM). This seemingly indolent condition was first described over 40 years ago after 6 patients who satisfied the diagnostic criteria for MM remained symptom free without myeloma-defining events (MDEs) after 5 years of follow-up.1 The clinical course for SMM is variable; some patients remain in the smoldering indolent phase for years, whereas other patients progress quickly to symptomatic MM. Given the heterogeneity within this group of patients, much has been done to better characterize and define SMM, understand underlying genomic drivers, and risk-stratify patients to identify those best served by early treatment approaches. In this article, we focus on updates in the diagnosis, risk stratification, and treatments for SMM.
Challenges in determining true epidemiology
The incidence of SMM increases with age.2,3 The median age of diagnosis is between 62 and 67 years.2,3 SMM is an uncommon entity, with an estimated standardized incidence between 0.4 and 0.9 cases per 100 000 people.2,4 In contrast, MGUS incidence is estimated to be 120 and 60 per 100 000 cases for men and women, respectively, by age 50 years, whereas MM incidence is 7.1 per 100 000 people.5,6
Challenges in accurately determining the epidemiology of SMM arise due to its rarity, underdiagnosis given its asymptomatic nature, and underrepresentation of minorities within public databases.7 In general, black people account for higher proportions of those afflicted at all stages of the monoclonal continuum, followed by Hispanic/LatinX people, whites, and those of east Asian descent.7-9 Furthermore, SMM lacks its own unique International Classification of Diseases diagnostic code and is instead considered under the diagnostic umbrella of MM.3 In a review of the National Cancer Database in the United States, 17% of patients with MM had a diagnosis of SMM.3
Refining and defining the diagnosis of SMM
The diagnosis of SMM has been refined over the years (Figure 1). Initially in 1980, SMM was defined as 10% or more of bone marrow (BM) PC infiltrate and/or monoclonal protein (M-protein) 3 g/dL or higher in the absence of any related end-organ impairment.1 The International Myeloma Working Group (IMWG) first formalized a consensus definition of SMM in 2003.13 However, further studies identified a subset of patients with a high clonal PC burden that rapidly progressed to MM. The presence of an elevated involved FLC ratio of 100 or higher or BM plasmacytosis of 60% or more increased risk of progression from smoldering to symptomatic MM to 72% and 95% in 2 years, respectively.11,15 In addition, MRI emerged to distinguish between smoldering and symptomatic MM; identification of 2 or more focal lesions on MRI correlated with rapid progression to symptomatic MM.12,14 Therefore, in 2014, the IMWG revised the MM definition to add “SLiM” (S: Sixty [60]% or more clonal plasma cells, Li: Light chains ratio involved to uninvolved >100, M: MRI >1 focal lesion on MRI) criteria (≥60% plasmacytosis, involved FLC ratio ≥100 and involved FLC ≥10 mg/dL, or >1 focal lesion on MRI) to include these “ultra-high-risk” patients with SMM.16 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)–CT is an established imaging modality for patients with SMM; diffuse FDG avidity without evidence of focal or osteolytic lesions is nondiagnostic for MM.17,18 Current SMM diagnostic criteria are shown in Figure 2.

Timeline leading to the current diagnosis and management of SMM. CRAB, C-hyperCalcemia, R-Renal impairment, A-Anemia, B-Bone lesions related to Multiple Myeloma.
Timeline leading to the current diagnosis and management of SMM. CRAB, C-hyperCalcemia, R-Renal impairment, A-Anemia, B-Bone lesions related to Multiple Myeloma.
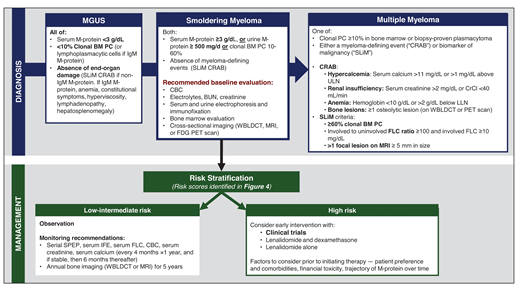
Diagnostic workup, risk stratification, and management of SMM. BUN, blood urea nitrogen; CBC, complete blood count; CrCl, creatinine clearance; IFE, immunofixation; LLN, lower limit of normal; SPEP, serum protein electrophoresis; ULN, upper limit of normal; WBLDCT, whole-body low-dose CT.
Diagnostic workup, risk stratification, and management of SMM. BUN, blood urea nitrogen; CBC, complete blood count; CrCl, creatinine clearance; IFE, immunofixation; LLN, lower limit of normal; SPEP, serum protein electrophoresis; ULN, upper limit of normal; WBLDCT, whole-body low-dose CT.
The role of genomics in understanding “who smolders longer”
Cytogenetic and genetic profiling of patients with SMM has provided insight into understanding the variable rates of progression.19 Primary cytogenetic events (trisomies and immunoglobulin heavy chain translocations) are inciting triggers of the aberrant PC in MGUS.20 However, the complexity of the genomic evolution from MGUS to MM is being studied with whole-exome and next-generation sequencing.19,21 Secondary genetic hits such as single-nucleotide variants of the mitogen-activated protein kinase pathway, DNA repair pathway alterations, MYC structural variants/dysregulation, copy number alterations, and translocations occur even at the smoldering stage, with aspects of the genomic architecture similar to MM (Figure 3).19,22-24 KRAS, Ig-MYC translocation, DNA pathway alterations, and APOBEC mutations are some of the genomic features associated with shorter time to MM progression.19,22,23 Two main patterns of clonal evolution have been elucidated to drive the progression of SMM.24 Patients with a “stable” pattern of evolution have a similar genomic landscape as they progress from SMM to MM; essentially, these patients have early MM and develop MDE as the tumor burden increases.24,25 In contrast, in patients with a “branching” evolutionary pattern, subclones change significantly as they progress from SMM to MM, and the time to progression (TTP) is longer because of the time required to acquire the genetic aberrations leading to overt MM.19,23-25 Epigenetic changes and contribution of the tumor microenvironment add further complexity to SMM progression.26 Dysregulated immune and cellular compartments are seen early in the MGUS phase.27 The immune aberrations continue at the SMM stage, where loss of memory T cells, decreased expression of activation and proliferation markers, and altered MHC II gene expression by CD14+ monocytes create an environment favoring cancer evasion.26-28
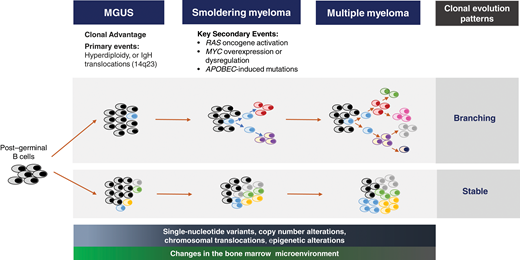
The evolutionary biology leading to SMM. The post–germinal B cell acquires a primary genetic defect at the MGUS stage, which triggers a dominant clone. The secondary genetic events that incite the transition to SMM include the development of chromosomal copy number alterations, translocations and single-nucleotide variants, and epigenetic changes. Some of the key high-risk secondary genomic events triggering the transition to SMM are RAS oncogene activation, MYC overexpression and dysregulation, and APOBEC-mediated mutations. Several theories are proposed for the clonal evolution from MGUS to MM. The dominant theory is the branching pattern of evolution, where generations of subclones develop from the parent clone. With stable clonal evolution, there is no major change in the clonal architecture throughout the monoclonal continuum to MM. Progressive changes in the stromal and cellular compartments of the bone marrow microenvironment facilitate expansion of the plasma cell clone and loss of immune surveillance.
The evolutionary biology leading to SMM. The post–germinal B cell acquires a primary genetic defect at the MGUS stage, which triggers a dominant clone. The secondary genetic events that incite the transition to SMM include the development of chromosomal copy number alterations, translocations and single-nucleotide variants, and epigenetic changes. Some of the key high-risk secondary genomic events triggering the transition to SMM are RAS oncogene activation, MYC overexpression and dysregulation, and APOBEC-mediated mutations. Several theories are proposed for the clonal evolution from MGUS to MM. The dominant theory is the branching pattern of evolution, where generations of subclones develop from the parent clone. With stable clonal evolution, there is no major change in the clonal architecture throughout the monoclonal continuum to MM. Progressive changes in the stromal and cellular compartments of the bone marrow microenvironment facilitate expansion of the plasma cell clone and loss of immune surveillance.
Assessing risk of progression to active MM
Risk stratification in SMM is particularly important to identify patients with SMM who benefit most from treatment, especially given potential treatment-related adverse events and financial implications. Clinical features associated with shorter time to MM include circulating PCs,29 PCs with an aberrant immune phenotype30 or a high proliferative index,31 focal lesions without osteolysis on PET imaging,17 cytogenetic markers such as deletion 17p or t(4;14),20 immunoparesis,30 and increased serum biomarkers. Common risk stratification models (Figure 4) have relied on surrogate measures of tumor burden for prognostication. The Mayo Clinic 2018 “20/20/2” model includes serum biomarkers (serum FLC ratio >20, serum monoclonal protein >2 g/dL) and BM PC burden more than 20% as risk factors.32 The IMWG validated the Mayo Clinic 2018 model with a cohort of 1996 patients, and the 2-year risk of progression to MM or amyloidosis in low-, intermediate-, and high-risk groups was 6%, 18%, and 44%, respectively.33 Risk factors in the older PETHEMA (Programa de Estudio y Tratamiento de las Hemopatías Maligna)model include the presence of immunoparesis and the percentage of PCs with an aberrant immunophenotype.30 However, the requirement for multiparameter flow cytometry makes the PETHEMA model difficult to implement clinically. Although the PETHEMA and Mayo Clinic 2018 models are used in clinical trials and practice, the classification of “high-risk” SMM is significantly discordant between the models.34 Therefore, to further optimize risk stratification, the IMWG recently developed a risk stratification model incorporating high-risk cytogenetic markers (t(4;14), t(14;16), gain 1q, monosomy 13/deletion 13q) and more refined criteria for risk factors included in the Mayo Clinic 2018 model.33 Using the IMWG 2020 model, intermediate- and high-risk patients with SMM had a 2-year risk of progression to myeloma or amyloidosis of 51% and 73%, respectively.33
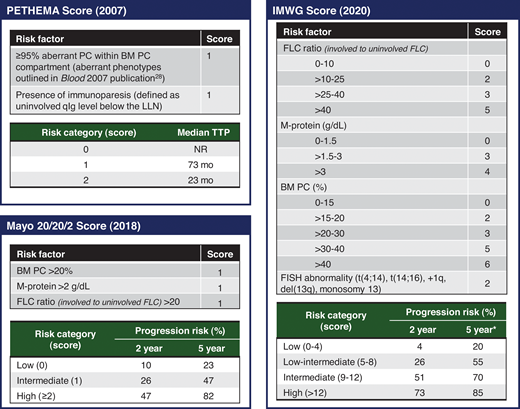
Commonly used risk stratification models in clinical practice. The risk factors and scoring PETHEMA, Mayo Clinic 2018, and IMWG 2020 scores are summarized, and the risk of progression based on scoring is outlined. FISH, fluorescence in situ hybridization; NR, not reached; PC, plasma cell; qIg, quantitative immunoglobulin. *The 5-year progression risk of the IMWG 2020 model is extrapolated from the Kaplan-Meier curve (figure 4 of the original publication).
Commonly used risk stratification models in clinical practice. The risk factors and scoring PETHEMA, Mayo Clinic 2018, and IMWG 2020 scores are summarized, and the risk of progression based on scoring is outlined. FISH, fluorescence in situ hybridization; NR, not reached; PC, plasma cell; qIg, quantitative immunoglobulin. *The 5-year progression risk of the IMWG 2020 model is extrapolated from the Kaplan-Meier curve (figure 4 of the original publication).
A significant limitation of the PETHEMA and Mayo Clinic 2018 models is that they are applied only at SMM diagnosis and assume progression risk remains constant over time. However, retrospective studies have shown that risk of progression is highest the first 5 years of SMM diagnosis and then stabilizes at 3% to 5% per year thereafter.32,35 To account for differing clinical trajectories of SMM, multiple groups have attempted to assess risk of progression based on the evolution of biomarkers over time,36-38 but these scores have not been robustly validated. Further work is needed to optimize and prospectively validate the risk stratification of SMM. Ideally, future risk models will incorporate dynamic changes in tumor burden with genetic markers that predict clonal biology and markers that characterize the permissive immune microenvironment.19,23,26
To treat and how to treat, those are the questions
Previously, SMM standard of care was observation until patients developed symptomatic MM. However, the advent of effective and safe treatments has challenged this status quo. Thus, questions about which patients with SMM to treat and how to treat are becoming increasingly relevant. Multiple early intervention strategies are being investigated: single vs combination therapy, lower intensity to delay progression to MM vs aggressive multiagent therapy with curative intent. A summary of published and ongoing phase 2 and 3 trials is summarized in Tables 1 and 2, respectively.
Summary of selected phase 2 and 3 studies within reported outcome data within the past decade
| Trial name/date | Study design | Criteria for defining SMM patient inclusion | Intervention (I) and control (C) arms | Median follow-up | Key outcomes |
|---|---|---|---|---|---|
| QUIREDEX Mateos et al (2013, updated 2016)39,40 | Phase 3 Randomized, open label | SMM (diagnosed <5 years) and either: • BM PC ≥10% and M-protein (IgG ≥3 g/dL, IgA ≥2 g/dL, Bence-Jones proteinuria >1 g/24 h) • BM PC ≥10% or M-protein (defined as above), with ≥95% aberrant PC and immunoparesis (≥1 uninvolved immunoglobulin >25% below LLN) | • I: Lenalidomide and dexamethasone (n = 57)—Lenalidomide 25 mg × 21/28 days for 9 cycles, then 10 mg × 21/28 days for a 2-year total duration. Dexamethasonase 20mg days 1-4 and days 12–15 of first 9 cycles and days 1-4 at biochemical progression. • C: Observation (n = 62) | 75 months* | Primary outcome—TTP (progression defined as end-organ damage) • Median TTP (I vs C): NR vs 23 months (HR, 0.24; 95% CI, 0.14-0.41) Secondary outcome—OS • Median OS (I vs C): NR in both groups (HR, 0.43; 95% CI, 0.21-0.92) |
| ECOG-ACRIN E3A06 Lonial et al (2019)10 | Phase 2/3 Randomized, open label | SMM (diagnosed <5 years) with ≥10% PCs and abnormal sFLC ratio (<0.26 or >1.65) | • I: Lenalidomide (n = 90)—25 mg (days 1-21 of 28 days), until progression or toxicity • C: Observation (n = 92) | 35 months | Primary outcome—PFS (progression defined as biochemical progression in addition to end-organ damage): • 3-year PFS (I vs C)—91% vs 66%, (HR, 0.28; 95% CI, 0.12-0.65) Additional outcomes (I vs C): • PFS in high-risk SMM subgroup (n = 56)—HR, 0.09 (95% CI, 0.02-0.44) • OS—HR, 0.46 (95% CI, 0.08-2.53) |
| CENTAURUS Landgren et al (2020)41 | Phase 2 Randomized, open label | SMM (diagnosed <5 years) with absence of SLiM or CRAB criteria and 1 of: • Serum M-protein ≥ 3 g/dL • iFLC/uFLC >8 if serum M-protein 1-3 g/dL • Urine M-protein >500 mg/24 h • Serum iFLC ≥100 mg/dL (if iFLC/uFLC between 8 and 99) | 3 arms based on daratumumab 16-mg/kg IV dosing schedule: • Intense (n = 41)— Q1W × 8, Q2W × 8, Q4W × 8, Q8W × 8 • Intermediate (n = 41)—Q1W × 8, Q8W × 19 • Short (n = 41)—Q1W × 8 | 25.8 months (prespecified primary analysis) | Co-Primary endpoint—≥ Complete response rate: • Intense arm—4.9% • Intermediate arm—9.8% • Short arm—0% Co-Primary endpoint—Progression† (progression defined as biochemical or end-organ damage) or death rate per patient year: • Intense arm—4.9% • Intermediate arm—9.8% • Short arm—0% |
| Trial name/date | Study design | Criteria for defining SMM patient inclusion | Intervention (I) and control (C) arms | Median follow-up | Key outcomes |
|---|---|---|---|---|---|
| QUIREDEX Mateos et al (2013, updated 2016)39,40 | Phase 3 Randomized, open label | SMM (diagnosed <5 years) and either: • BM PC ≥10% and M-protein (IgG ≥3 g/dL, IgA ≥2 g/dL, Bence-Jones proteinuria >1 g/24 h) • BM PC ≥10% or M-protein (defined as above), with ≥95% aberrant PC and immunoparesis (≥1 uninvolved immunoglobulin >25% below LLN) | • I: Lenalidomide and dexamethasone (n = 57)—Lenalidomide 25 mg × 21/28 days for 9 cycles, then 10 mg × 21/28 days for a 2-year total duration. Dexamethasonase 20mg days 1-4 and days 12–15 of first 9 cycles and days 1-4 at biochemical progression. • C: Observation (n = 62) | 75 months* | Primary outcome—TTP (progression defined as end-organ damage) • Median TTP (I vs C): NR vs 23 months (HR, 0.24; 95% CI, 0.14-0.41) Secondary outcome—OS • Median OS (I vs C): NR in both groups (HR, 0.43; 95% CI, 0.21-0.92) |
| ECOG-ACRIN E3A06 Lonial et al (2019)10 | Phase 2/3 Randomized, open label | SMM (diagnosed <5 years) with ≥10% PCs and abnormal sFLC ratio (<0.26 or >1.65) | • I: Lenalidomide (n = 90)—25 mg (days 1-21 of 28 days), until progression or toxicity • C: Observation (n = 92) | 35 months | Primary outcome—PFS (progression defined as biochemical progression in addition to end-organ damage): • 3-year PFS (I vs C)—91% vs 66%, (HR, 0.28; 95% CI, 0.12-0.65) Additional outcomes (I vs C): • PFS in high-risk SMM subgroup (n = 56)—HR, 0.09 (95% CI, 0.02-0.44) • OS—HR, 0.46 (95% CI, 0.08-2.53) |
| CENTAURUS Landgren et al (2020)41 | Phase 2 Randomized, open label | SMM (diagnosed <5 years) with absence of SLiM or CRAB criteria and 1 of: • Serum M-protein ≥ 3 g/dL • iFLC/uFLC >8 if serum M-protein 1-3 g/dL • Urine M-protein >500 mg/24 h • Serum iFLC ≥100 mg/dL (if iFLC/uFLC between 8 and 99) | 3 arms based on daratumumab 16-mg/kg IV dosing schedule: • Intense (n = 41)— Q1W × 8, Q2W × 8, Q4W × 8, Q8W × 8 • Intermediate (n = 41)—Q1W × 8, Q8W × 19 • Short (n = 41)—Q1W × 8 | 25.8 months (prespecified primary analysis) | Co-Primary endpoint—≥ Complete response rate: • Intense arm—4.9% • Intermediate arm—9.8% • Short arm—0% Co-Primary endpoint—Progression† (progression defined as biochemical or end-organ damage) or death rate per patient year: • Intense arm—4.9% • Intermediate arm—9.8% • Short arm—0% |
Median follow-up for surviving patients.40
Progression was defined based on the IMWG 2014 diagnostic criteria for MM, as well as the IMWG FLC progression criteria (a ≥25% increase from nadir in the difference between involved and uninvolved FLC with absolute increase >10 mg/dL).
C, control arm; CRAB, C-hyperCalcemia, R-Renal impairment, A-Anemia, B-Bone lesions related to multiple myeloma; iFLC, involved FLC; I, intervention arm; IV, intravenous; LLN, lower limit of normal; NR, not reached; sFLCr, serum FLC ratio; uFLC, uninvolved FLC.
Active or planned phase 2 and 3 studies for intermediate- and high-risk SMM
| Study (clinicaltrials.gov identifier) | Phase | Estimated enrollment | Recruitment status | Estimated study completion date | Interventions | Primary end point | Preliminary efficacy data reported |
|---|---|---|---|---|---|---|---|
| NCT04270409 | Phase 3 Randomized | 300 | Recruiting | 2033 | • Intervention arm: Isatuximab + Rd • Control arm: Rd | PFS | — |
| DETER-SMM NCT03937635 | Phase 3 Randomized | 288 | Recruiting | 2028 | • Intervention arm: DRD • Control arm: Rd • Both arms treated for up to 24 cycles (in the absence of disease progression or unacceptable toxicity) | OS and FACT- G score (quality-of-life measure) | — |
| AQUILA NCT03301220 | Phase 3 Randomized | 390 | Active, not recruiting | 2025 | • Intervention: subcutaneous daratumumab • Control: observation | PFS | — |
| NCT03850522 | Phase 2a Single arm | 20 | Recruiting | 2021 | • PD-L1 peptide vaccination subcutaneously every 2 weeks (total 26-week treatment duration) | ORR (≥PR) | — |
| NCT03839459 | Phase 2 Single arm | 20 | Recruiting | 2024 | • Subcutaneous Denosumab every 4 weeks | Reduction in SMM risk category | — |
| ASCENT NCT03289299 | Phase 2 Single arm | 83 | Recruiting | 2026 | • D-KRD × 6 cycles (induction) • D-KRD × 6 cycles (consolidation) • DR × 12 cycles (maintenance) | Stringent CR at any point during treatment | (Only safety data reported to date) |
| HO147SMM NCT03673826 | Phase 2 Randomized | 120 | Recruiting | 2025 | • Intervention arm: KRD × 9 cycles → R alone (up to 24 cycles) • Control arm: Rd × 9 cycles → R alone (up to 24 cycles) | PFS | — |
| NCT04775550 | Phase 2 Single arm | 30 | Not yet recruiting | 2026 | • D-VRD × up to 24 cycles (in absence of disease progression or toxicity) | 2-y MRD−rate | — |
| NCT04776395 | Phase 2 | 68 | Not yet recruiting | 2023 | • Arm A: Iberdomide + dexamethasone × 4 cycles (induction) → Iberdomide alone until disease progression or unacceptable toxicity • Arm B: Iberdomide alone until disease progression or unacceptable toxicity | ORR (≥PR) | — |
| E-PRISM NCT0227939442 | Phase 2 Single arm | 51 | Active, not recruiting | 2023 | • Elotuzumab + Rd × 8 cycles (induction) → Elotuzumab + R × cycles 9-24 (maintenance) | PFS | • Median follow-up not reported (n = 50) • PFS data NR • ORR 84%, CR 6% |
| NCT0291677143 | Phase 2 Single arm | 55 | Active, not recruiting | 2024 | • Ixazomib + Rd × 9 cycles (induction) → Ixazomib + R cycles 10-24 (maintenance) | PFS | • Median 8 cycles completed (n = 26) • No progression to date • ORR 89%, CR 19% |
| NCT0296055544 | Phase 2 Single arm | 61 | Active, not recruiting | 2022 | • Intervention: isatuximab IV × up to 30 cycles (in absence of disease progression or toxicity) | ORR (≥PR) | • Median 11.5 cycles completed (n = 24) • ORR 62.5% |
| GEM-CESAR NCT0241541345,46 | Phase 2 Single arm | 90 | Active, not recruiting | 2027 | • KRD × 6 cycles (induction) → melphalan conditioning and ASCT (intensification) → KRD × 2 cycles (consolidation) → Rd × 2 years (maintenance) | MRD−NGF (next generation flow) postinduction and ASCT | • Median follow-up 32 months (n = 90) • MRD−: 30% postinduction, 52% post-ASCT, 57% postconsolidation • MRD− and ≥CR: 23% postinduction, 44% post-ASCT, 55% postconsolidation |
| NCT0157248047 | Phase 1/2 Single arm | 52 | Active, not recruiting | 2025 | • Phase 1: KRD × 8 cycles (induction) → R alone for 12 cycles (maintenance) • Phase 2: KRD × 8 cycles (induction) → R alone for up to 24 cycles (maintenance) | MRD−CR (NGF, ≤10–5 sensitivity) | • Median follow-up 27.3 months (n = 52) • MRD−CR: 70.2 months |
| Study (clinicaltrials.gov identifier) | Phase | Estimated enrollment | Recruitment status | Estimated study completion date | Interventions | Primary end point | Preliminary efficacy data reported |
|---|---|---|---|---|---|---|---|
| NCT04270409 | Phase 3 Randomized | 300 | Recruiting | 2033 | • Intervention arm: Isatuximab + Rd • Control arm: Rd | PFS | — |
| DETER-SMM NCT03937635 | Phase 3 Randomized | 288 | Recruiting | 2028 | • Intervention arm: DRD • Control arm: Rd • Both arms treated for up to 24 cycles (in the absence of disease progression or unacceptable toxicity) | OS and FACT- G score (quality-of-life measure) | — |
| AQUILA NCT03301220 | Phase 3 Randomized | 390 | Active, not recruiting | 2025 | • Intervention: subcutaneous daratumumab • Control: observation | PFS | — |
| NCT03850522 | Phase 2a Single arm | 20 | Recruiting | 2021 | • PD-L1 peptide vaccination subcutaneously every 2 weeks (total 26-week treatment duration) | ORR (≥PR) | — |
| NCT03839459 | Phase 2 Single arm | 20 | Recruiting | 2024 | • Subcutaneous Denosumab every 4 weeks | Reduction in SMM risk category | — |
| ASCENT NCT03289299 | Phase 2 Single arm | 83 | Recruiting | 2026 | • D-KRD × 6 cycles (induction) • D-KRD × 6 cycles (consolidation) • DR × 12 cycles (maintenance) | Stringent CR at any point during treatment | (Only safety data reported to date) |
| HO147SMM NCT03673826 | Phase 2 Randomized | 120 | Recruiting | 2025 | • Intervention arm: KRD × 9 cycles → R alone (up to 24 cycles) • Control arm: Rd × 9 cycles → R alone (up to 24 cycles) | PFS | — |
| NCT04775550 | Phase 2 Single arm | 30 | Not yet recruiting | 2026 | • D-VRD × up to 24 cycles (in absence of disease progression or toxicity) | 2-y MRD−rate | — |
| NCT04776395 | Phase 2 | 68 | Not yet recruiting | 2023 | • Arm A: Iberdomide + dexamethasone × 4 cycles (induction) → Iberdomide alone until disease progression or unacceptable toxicity • Arm B: Iberdomide alone until disease progression or unacceptable toxicity | ORR (≥PR) | — |
| E-PRISM NCT0227939442 | Phase 2 Single arm | 51 | Active, not recruiting | 2023 | • Elotuzumab + Rd × 8 cycles (induction) → Elotuzumab + R × cycles 9-24 (maintenance) | PFS | • Median follow-up not reported (n = 50) • PFS data NR • ORR 84%, CR 6% |
| NCT0291677143 | Phase 2 Single arm | 55 | Active, not recruiting | 2024 | • Ixazomib + Rd × 9 cycles (induction) → Ixazomib + R cycles 10-24 (maintenance) | PFS | • Median 8 cycles completed (n = 26) • No progression to date • ORR 89%, CR 19% |
| NCT0296055544 | Phase 2 Single arm | 61 | Active, not recruiting | 2022 | • Intervention: isatuximab IV × up to 30 cycles (in absence of disease progression or toxicity) | ORR (≥PR) | • Median 11.5 cycles completed (n = 24) • ORR 62.5% |
| GEM-CESAR NCT0241541345,46 | Phase 2 Single arm | 90 | Active, not recruiting | 2027 | • KRD × 6 cycles (induction) → melphalan conditioning and ASCT (intensification) → KRD × 2 cycles (consolidation) → Rd × 2 years (maintenance) | MRD−NGF (next generation flow) postinduction and ASCT | • Median follow-up 32 months (n = 90) • MRD−: 30% postinduction, 52% post-ASCT, 57% postconsolidation • MRD− and ≥CR: 23% postinduction, 44% post-ASCT, 55% postconsolidation |
| NCT0157248047 | Phase 1/2 Single arm | 52 | Active, not recruiting | 2025 | • Phase 1: KRD × 8 cycles (induction) → R alone for 12 cycles (maintenance) • Phase 2: KRD × 8 cycles (induction) → R alone for up to 24 cycles (maintenance) | MRD−CR (NGF, ≤10–5 sensitivity) | • Median follow-up 27.3 months (n = 52) • MRD−CR: 70.2 months |
ASCT, autologous stem cell transplant; D-KRD, daratumumab, carfilzomib, lenalidomide, and dexamethasone; DRD, daratumumab, lenalidomide, and dexamethasone; D-VRD, daratumumab, lenalidomide, dexamethasone; FACT-G, functional assessment of cancer therapy-general; IV, intravenous; ORR, overall response rate; NGF, next generation flow; PR, partial response.
The first phase 3 trial for SMM was the QUIREDEX trial, which randomized 119 high-risk patients with SMM to either lenalidomide and dexamethasone (Rd) vs observation.39,40 Patients in the intervention arm received 9 cycles of Rd induction followed by lenalidomide maintenance, for a total treatment duration of 2 years. At a median follow-up of 75 months, the time to progression to symptomatic MM was not reached with early intervention compared with 23 months with observation (hazard ratio [HR], 0.24; 95% CI, 0.14-0.41).40 Overall survival (OS), a secondary end point, was also significantly longer with early therapeutic intervention compared with observation (HR, 0.43; 95% CI, 0.21-0.92). Although survival data are encouraging, a few caveats need to be considered. The QUIREDEX trial was conducted prior to the update in MM diagnostic criteria, so cross-sectional imaging and evaluation of FLCs were not required at enrollment. Therefore, patients meeting the current criteria for MM may have been included in this study, which may explain the early progression in the observation arm. Some experts have raised concerns about early intervention leading to selection of resistant PC clones. Interestingly, a post hoc analysis of QUIREDEX patients showed a similar OS from the time of active MM between patients initially randomized to early intervention vs observation strategies, suggesting that intervention did not lead to development of treatment-resistant clones.
The ECOG E3A06 phase 3 randomized trial was conducted to see if the immunomodulating effects of lenalidomide could delay progression to active myeloma without additional corticosteroids.10 In this trial, 182 patients with SMM were randomized to lenalidomide monotherapy (given until disease progression) or observation. Patient accrual began in 2013, thereby including a subset of patients who satisfied the later updated 2014 criteria for MM (3.3% patients had >60% BM PC involvement, and 8.2% had FLC ratio >100). Unlike the QUIREDEX trial, an MRI of the spine and pelvis was performed for all eligible patients. At a median follow-up of 35 months, the progression-free survival (PFS) was significantly longer with lenalidomide intervention compared with observation (3-year PFS 91% vs 66%, respectively; HR, 0.28; 95% CI, 0.12-0.62). Importantly, progression was defined as the development of end-organ dysfunction and did not include “SLiM” criteria. The improved PFS with lenalidomide was observed primarily in the high-risk SMM subgroup, which constituted 56 (38%) patients overall (27.8% vs 33.7% of patients in the lenalidomide and observation arms, respectively), with an HR of 0.09 (95% CI, 0.02-0.44). Importantly, the PFS improvement did not reach statistical significance in low- to intermediate-risk SMM subgroups. The most common MDEs were bone lesions or soft tissue plasmacytomas (n = 14 total, 50% of progression events), followed by anemia (n = 12, 42.9% of progression events). Whether patients were symptomatic at the time of progression is unknown, which is relevant because treatment of asymptomatic anemia or bone lesions may not be as clinically meaningful to patients who are otherwise well. OS data were not mature at the time of publication, and few deaths had occurred. Therefore, it is currently unclear if early intervention with lenalidomide results in a survival benefit. Although the study was designed to continue lenalidomide until progression or unacceptable toxicity, and there was no difference in health-related quality of life between study arms, the median treatment duration was 23 months with 40% of lenalidomide-treated patients discontinuing therapy due to adverse effects. Grade 3 to 4 hematologic and nonhematologic adverse events occurred in 41% and 28% of treated patients, respectively. Thus, study authors suggested treatment be limited to 2 years total. In an otherwise asymptomatic patient population, weighing the risks of treatment-related toxicity against the benefit of prolonging time to symptomatic disease needs to be carefully scrutinized, especially in the absence of OS data.
Carfilzomib, lenalidomide, and dexamethasone (KRD)–based early-intervention approaches with reported preliminary data have shown deep responses; the GEM-CESAR trial reported a postconsolidation minimal residual disease negative (MRD–) rate of 57%, and a nontransplant approach showed an MRD– complete response (CR) rate of 70%.45,47 Although these responses are promising, the utility of an MRD end point and impact of acute and long-term toxicities of intensive therapy remain to be seen.
Given what we know, what is the standard of care for SMM?
Presently, for low-risk SMM, the standard of care is active surveillance. The IMWG recommends that patients be monitored every 4 months for a year to evaluate the trajectory of biomarkers, and if stable, evaluations can be increased to 6-month intervals.
Currently, there remains no clear consensus regarding optimal management of high-risk SMM. Given the evolving definition of MM and heterogeneity of defining high-risk SMM between trials, cross-trial comparison in SMM studies is challenging. Until there are clear evidence-based guidelines, our approach to managing high-risk SMM is to recommend clinical trial enrollment, if available. Based on significant PFS improvements in high-risk patients with SMM and the morbidity associated with active MM, we advocate for early therapeutic intervention in high-risk patients (those with a 2-year progression risk approximately >50%). The current debate relates to how these patients should be treated. We suggest that high-risk patients with SMM with a clearly “evolving” clinical phenotype (increasing M-proteins over a short observation period, shown in Figure 5, trajectory “B”) should be treated as early MM or with regimens studied in high-risk SMM phase 3 trials (either fixed-duration Rd or lenalidomide alone) and collecting and storing autologous stem cells after 4 to 6 cycles of therapy. However, for high-risk patients with SMM with stable M-proteins (Figure 5, trajectory “A”), we recommend lenalidomide with or without dexamethasone. In all treatment decisions, patient preferences and comorbidities should be carefully evaluated before initiation.
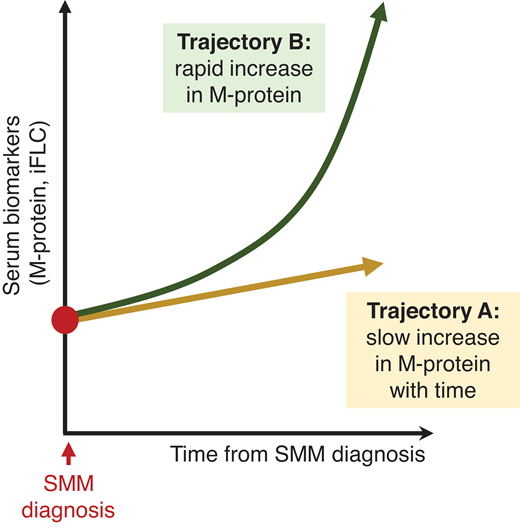
Hypothetical changes in the monoclonal protein markers of patients with SMM over time. While there are multiple possible trajectories that a patient's biomarkers may take, this figure is meant to illustrate that patients may have steady increases in the tumor burden, whereas other patients have a rapidly “evolving” presentation. Patients in “trajectory B,” with rapidly increasing biomarkers, should be considered for early therapeutic intervention.
Hypothetical changes in the monoclonal protein markers of patients with SMM over time. While there are multiple possible trajectories that a patient's biomarkers may take, this figure is meant to illustrate that patients may have steady increases in the tumor burden, whereas other patients have a rapidly “evolving” presentation. Patients in “trajectory B,” with rapidly increasing biomarkers, should be considered for early therapeutic intervention.
Future of SMM
With strides made in defining the genomic landscape of SMM and advances in refining risk stratification, the future of SMM is bright. Still, many questions remain unanswered, such as “Is SMM in fact just early myeloma?” or “Should all SMMs be treated?” We anticipate that future prediction and risk stratification criteria will focus less on tumor burden and more on genomic architecture, clonal patterns of progression, and epigenetic alterations as prognostic markers. Results from ongoing trials of innovative treatment combinations and novel therapeutic approaches in high-risk SMM will provide clarity and transform the treatment landscape in the near future.
CLINICAL CASE (continued)
Our patient has 2 risk factors (M-protein >2 g/dL and BM PC >20%) and is “high risk” according to the Mayo Clinic 2018 model. Given the high risk of progression to MM, we discussed the risks and benefits of beginning treatment with Rd. Fifteen months later, he remains progression free, experiencing minimal side effects.
Conflict-of-interest disclosure
Alissa Visram: no conflicts to disclose.
Joselle Cook: no conflicts to disclose.
Rahma Warsame: no conflicts to disclose.
Off-label drug use
Alissa Visram: None of the medications are FDA approved; all that are discussed are off label.
Joselle Cook: None of the medications are FDA approved; all that are discussed are off label.
Rahma Warsame: None of the medications are FDA approved; all that are discussed are off label.
