
Waldenström macroglobulinemia (WM) is a neoplasm of small B lymphocytes, plasmacytoid lymphocytes, and plasma cells typically involving the bone marrow, lymph nodes, and spleen. It is actually a subcategory of the disease entity known at lymphoplasmacytic lymphoma (LPL). WM is defined as LPL with marrow involvement and an IgM paraprotein of any concentration; most LPLs will qualify as WM. Hyperviscosity, anemia, and neuropathy are some of the more common clinical manifestations. The goal of therapy for this incurable, indolent neoplasm is disease control, with good quality of life, to avoid these clinical complications. More than 90 percent of WM cases have the activating MYD88 L265P mutation, which increases intracellular signaling through Bruton tyrosine kinase (BTK), making BTK a rational target for therapy. This mutation is so prevalent in WM, that it can be used as a diagnostic aid. However, another 30 to 40 percent of WM cases will have activating mutations in CXCR4, and these mutations appear to confer some resistance to BTK inhibition. So how does one integrate this information when selecting therapy in WM?
As of this writing, ibrutinib is the sole BTK inhibitor approved by the U.S. Food and Drug Administration (FDA) in WM, and it is approved in both the frontline and the relapsed/refractory (R/R) settings. Dr. Steven P. Treon and colleagues first reported on the efficacy of the BTK inhibitor ibrutinib for WM in 2015.1 Recently, they provided the five-year follow-up to this study. The results are informative and should influence clinical decision-making. The long-term follow-up includes 63 patients with previously treated WM, all receiving ibrutinib at a starting dose of 420 mg daily. The treatment was generally well tolerated with rare grade 3-4 adverse events. Eight patients developed atrial arrythmias, but seven of those patients were able to stay on ibrutinib therapy with successful medical management of their persistent arrythmia. The major response rate for the entire group was 80 percent, and the five-year progression-free survival (PFS) rate for the entire group was 54 percent. When evaluating response by mutational status some significant differences emerge (Table).
Outcomes Based on Mutational Profiling
| Subtype | No. of Patients | Major Response | 5-year PFS |
|---|---|---|---|
| MYD88Mut/CXCR4WT | 36 | 97% | 70% |
| MYD88Mut/CXCR4Mut | 22 | 68% | 38% |
| MYD88WT/CXCR4WT | 4 | 0% | 0% |
| Subtype | No. of Patients | Major Response | 5-year PFS |
|---|---|---|---|
| MYD88Mut/CXCR4WT | 36 | 97% | 70% |
| MYD88Mut/CXCR4Mut | 22 | 68% | 38% |
| MYD88WT/CXCR4WT | 4 | 0% | 0% |
PFS, progression-free survival.
Clearly, the patients in group 1 experienced the best results from single-agent ibrutinib therapy. If you discover this pattern, ibrutinib is a logical choice. The group 3 patients (note that MYD88WT is rare in WM) derive little benefit from ibrutinib, and one is better served trying other strategies. But what to do with group 2 patients, who seem to derive an intermediate result? I would argue that ibrutinib is still a very good choice for these patients. Looking a bit deeper at the data reveals why. When one adds minor responses (decrease in serum IgM levels by 25-49%), the overall response rate in group 2 patients jumps to 86 percent. Minor responses to ibrutinib are typically associated with good improvement in the hemoglobin levels, so the clinical benefit to the patient is significant. The median PFS in the group 2 patients was 4.5 years (Figure), which while not as durable as the group 1 PFS, is still a very good result.
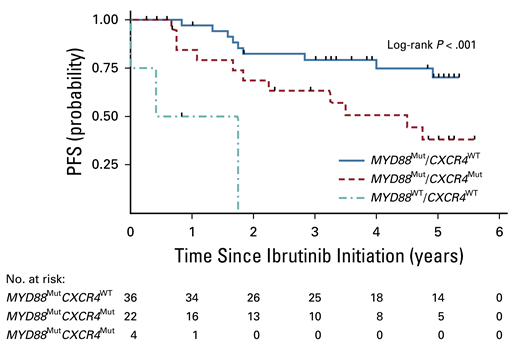
Progression-free survival (PFS) based on mutational profiling. Reprinted with permission. © 2021 American Society of Clinical Oncology. All rights reserved. Treon SP et al: J Clin Oncol Vol. 39 (Issue 6), 2021: 565-575.
Progression-free survival (PFS) based on mutational profiling. Reprinted with permission. © 2021 American Society of Clinical Oncology. All rights reserved. Treon SP et al: J Clin Oncol Vol. 39 (Issue 6), 2021: 565-575.
In Brief
My take-home is that any patient with WM with an MYD88 mutation, which is more than 90 percent of the population, is a good candidate for ibrutinib therapy in the R/R setting. Additional good news may be on the way, pending FDA approval. Zanubrutinib, a more selective BTK inhibitor, has demonstrated comparable clinical activity with a better safety and tolerability profile compared to ibrutinib in R/R WM.2
Competing Interests
Dr. Kahl reports consulting fees from Pharmacyclics, Abbvie, Janssen, and BeiGene.
