

Abstract
Abstract 160
Paroxysmal nocturnal hemoglobinuria (PNH) is characterized by hemolytic anemia, bone marrow failure and venous thromboembolism. The disease is caused by a somatic mutation of the X-linked gene PIG-A, encoding a key enzyme responsible for the biosynthesis of the glycosylphosphatidylinositol anchored proteins (GPI-APs). Unfolded GPI-APs are translated and degraded intracellularly in the endoplasmic reticulum (ER) via the proteasome machinery. We hypothesized that the accumulation of misfolded proteins in mutant PNH cells activates the unfolded protein response (UPR) signaling cascade, which physiologically maintains the quality of newly synthesized proteins. UPR could thus represent a new target for therapy in PNH. We first assessed the sensitivity of GPI-AP- deficient K562 cells to the proteasome inhibitor PS-341 (Velcade‘) by flow cytometry after staining for annexin V and propidium iodide (PI). After 24 hour exposure to PS-341, cytotoxicity was significantly higher in the K562 GPI-AP-deficient cell line compared to their wild type counterpart at all dose tested (0 to 200nM). At 50nM, the early stage apoptosis was 21±2% in the K562 GPI-AP-deficient cell line versus 9.3±2% for their wild type counterpart (p=0.013) while the late stage apoptosis was 38±2.3% versus 22±2.3% (p<10−3), respectively. Similar results were obtained in immortalized lymphoblastoid cells derived from a PNH patient after PS-341 exposure. We reproduced those results with 2 other proteasome inhibitors (MG132 and Lactacystine) confirming the fundamental role of the proteasome in PNH. By immunoblot, K562 GPI-AP-deficient cell line exhibited more expression of chaperone proteins (CHOP and BiP/GRP78) compared to wild type cells after PS-341 exposure; evidence of the UPR is initiated by accumulation of unfolded proteins. UPR activation was confirmed by the detection of XBP1 splicing form in K562 GPI-AP-deficient cells by polymerase chain reaction (PCR). Moreover, PS-341 promoted pro-apoptotic/terminal UPR gene expression in K562 GPI-APs deficient cells as illustrated by the increase expression of an UPR specific pro-apoptotic protein, NOXA. We observed a concomitant arrest in the G2 phase of the cell cycle, as detected by flow cytometry after staining with PI. We employed a mouse model bearing a conditional Pig-a gene deletion in hematopoietic cells. When the animals were treated with PS-341 (15 ug/day once a week, intraperitoneally), there was a significantly higher proportion of apoptotic GPI-AP-deficient B cells compared to normal B cells in early stage apoptosis (p=0.01) and a slight increase of in the late stage apoptosis (p=0.13). PS-341 induced apoptosis also in bone marrow samples from two patients with classical and aplastic PNH as assessed by flow cytometry, based on CD59 and CD55 expression in the CD34+ population (Figure 1). GPI-AP-CD34+ deficient cells were more sensitive to PS-341 at the early stage of apoptosis (at 50 nM, 14% vs 4.6%), as well as late stage (at 200nM: 22% vs 7.2%). In conclusion, we demonstrate that GPI-AP-deficient cells are more susceptible to proteasome inhibition through the activation of the proapoptotic/terminal UPR, which leads to specific apoptosis in these cells. The sensitivity of PNH cells in vivo and in vitro to proteasome inhibitors might allow for the development of therapeutic strategies specifically directed to target one or several components of the UPR in PNH.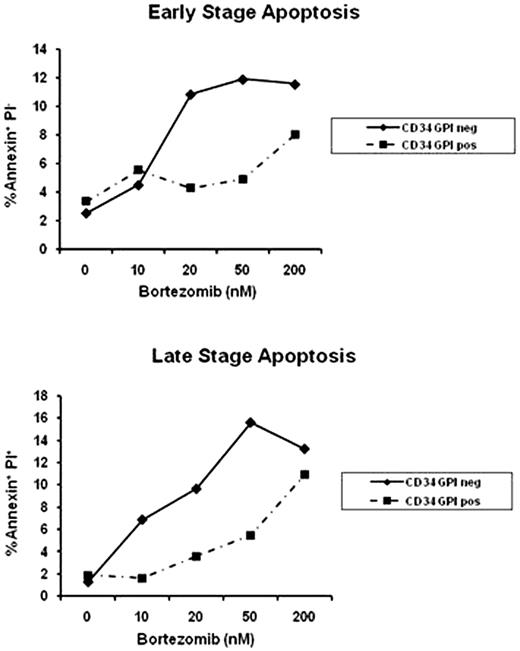 Close modal
Close modal
Figure 1:

Representative apoptosis assay in CD34+ cells according to GPI expression after 24 hours exposition to PS-341
Figure 1:
Representative apoptosis assay in CD34+ cells according to GPI expression after 24 hours exposition to PS-341
Disclosures:
No relevant conflicts of interest to declare.

