

Human T-lymphotropic virus type 1 (HTLV-1) spreads directly between lymphocytes and other cells via a specialized cell-cell contact, termed the virological synapse. The formation of the virological synapse is accompanied by the orientation of the microtubule-organizing center (MTOC) in the infected T cell toward the cell contact region with the noninfected target cell. We previously demonstrated that the combination of intracellular Tax protein expression and the stimulation of the intercellular adhesion molecule-1 (ICAM-1) on the cell surface is sufficient to trigger MTOC polarization in the HTLV-1–infected T cell. However, the mechanism by which Tax and ICAM-1 cause the MTOC polarization is not fully understood. Here we show that the presence of Tax at the MTOC region and its ability to stimulate cyclic AMP-binding protein–dependent pathways are both required for MTOC polarization in the HTLV-1–infected T cell at the virological synapse. Furthermore, we show that the MTOC polarization induced by ICAM-1 engagement depends on activation of the Ras-MEK-ERK signaling pathway. Our findings indicate that efficient MTOC polarization at the virological synapse requires Tax-mediated stimulation of T-cell activation pathways in synergy with ICAM-1 cross-linking. The results also reveal differences in the signaling pathways used to trigger MTOC polarization between the immunologic synapse and the virological synapse.
Introduction
Human T-lymphotropic virus type 1 (HTLV-1) is the etiologic agent of adult T-cell leukemia/lymphoma1 and is also associated with several inflammatory disorders, including HTLV-1–associated myelopathy/tropical spastic paraparesis.2,3 Naturally infected lymphocytes produce virtually no cell-free virions in vivo, and cell contact is required for efficient transmission of HTLV-1 between cells and between persons. Dendritic cells may be infected with cell-free HTLV-1 particles, but transmission from dendritic cells to T cells also requires cell contact.4 We previously showed that when an HTLV-1–infected T cell meets an uninfected T cell, the microtubule organizing center (MTOC) of the infected cell becomes polarized toward the uninfected cell. Complexes of HTLV-1 core protein (Gag) and the RNA genome of the virus accumulate at the point of cell contact and are then transferred to the uninfected cell.5
Microtubule polarization plays a central part in cell migration,6,7 and in effector responses by CD4+ T cells,8 natural killer (NK) cells, and cytotoxic T lymphocytes.9,10 Rapid MTOC reorientation to the cell contact area is accompanied by other cellular components, such as Golgi apparatus9,11 or cytotoxic granules.12
Adhesion molecules present at the cell surface, in particular ICAM-1 and its ligand LFA-1, play a key role at the early stage of the formation of the virological synapse. We previously showed that the engagement of these molecules is sufficient to trigger the reorientation of the MTOC in HTLV-1–infected T cells toward the cell-cell contact region.13,14 Others have shown that the expression of LFA-1 at the surface of the target cell is essential in the cell-cell transmission of HIV-1.15,16 Furthermore, it has been shown that the expression of HTLV-1 accessory protein p12 modulates the expression of LFA-1 and ICAM-1 on the surface of the infected T cell.17,18
Tax is among the first HTLV-1 proteins to be translated during ex vivo culture of infected peripheral blood mononuclear cells (PBMCs).19 Tax protein plays a critical role in cellular transformation20,–22 ; it activates the expression both of HTLV-1 genes through the viral long terminal repeat and several cellular genes through cellular signaling pathways of nuclear factor-κB (NF-κB), cyclic-AMP response element-binding protein (CREB), serum response factor (SRF), and the transcription factor complex AP-1. AP-1 plays a major role in the regulation of proliferation and activation of T cells and in cytokine production.23,24 In nonstimulated normal T cells, the basal level of AP-1 proteins is low, but T-cell activation results in rapid induction of jun and fos genes.25 AP-1 activity is also regulated by the activation of c-Jun N-terminal kinase (JNK).26 JNK phosphorylates c-Jun, thereby increasing its DNA-binding activity.27 HTLV-1 infection activates this pathway by inducing the expression of various members of the AP-1 family, including c-Jun, and by constitutively activating the ERK-JNK cascade. However, this activation is not induced by Tax alone.24,28,–30 In addition, recent reports showed that activation of mitogen-activated protein kinase (MAPK) pathways, especially ERK and JNK, is also required for MTOC and cytotoxic granule polarization in human NK cells when NKG2D is ligated.31,–33
Tax acts on genes indirectly, by binding several transcription factors, including NF-κB, CREB/activated transcription factor, and CREB-binding protein CBP/p300. Tax protein is subjected to several posttranscriptional modifications, including phosphorylation,34 and ubiquitination or sumoylation at each of several lysine residues.35 The ubiquitinated forms of Tax protein were not targeted for proteasomal degradation22 ; rather, it appeared that the ubiquitination of Tax protein was required for its ability to activate the NF-κB pathway.36,37
We previously reported that Tax protein expression is sufficient to account for the strong polarization of the MTOC associated with the formation of the HTLV-1 viral synapse. We postulated that there is a synergy between intracellular expression of Tax protein and the engagement of the ICAM-1 on the cell surface to induce MTOC polarization in HTLV-1–infected cells.13,14 In the present study, using a range of Tax mutants deficient in specific Tax functions, we investigated the mechanisms by which Tax protein induces MTOC polarization in the T-lymphocyte.
Methods
Antibodies and reagents
Monoclonal antibodies against HTLV-1 proteins Tax (Lt-4) and GagP19 (GIN7)14,38 included anti–β-tubulin (TUB2.1) CY3-conjugated and anti–γ-tubulin (Sigma-Aldrich), anti–ICAM-1 (HA58) (Research Diagnostics), and anti-CD3 (HIT3a; Insight Biotechnology).
Cycloheximide, nocodazole, and cytochalasin B (Sigma-Aldrich) were used at a final concentration of 10, 33, and 40 μM, respectively. To inhibit MAP-kinase kinase (MEK), we used the drugs PD98059 (2′-amino-3′-methoxyflavone) and U0126 (2′ 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio) butadiene at a final concentration of 20 μM and 100 μM, respectively. STO-609 (an ATP-competitive inhibitor of Ca2+/calmodulin-dependent protein-kinase kinase) was used at a final concentration of 10 μM. These kinase inhibitors were purchased from Calbiochem (Merck Biosciences).
HTLV-1–infected T cells and cell lines
PBMCs were obtained from patients with a high proviral load attending the National Center for Human Retrovirology at St Mary's Hospital (London, United Kingdom) under protocols approved by the Institutional Review Board of Imperial College London. All patients gave written informed consent in accordance with the Declaration of Helsinki. HTLV-1–infected CD4+ T cells were separated from the PBMCs using antibody-coupled magnetic beads and cultured as previously described.19 Jurkat E6.1 (ATCC) is a clone derived from the human leukemic T-cell line Jurkat.39 The HTLV-1–producing cell line (MS-9), a gift of Dr David Derse (National Cancer Institute, National Institutes of Health) was derived by coculture of human PBMCs with DBS-FRhL (clone B5) cells infected with the HTLV-1 molecular clone, pHTLV-X1MT.40 The cells were cultured in RPMI 1640 (Sigma-Aldrich) containing 2 mM glutamine (Invitrogen), 50 IU/mL penicillin, and 50 μg/mL streptomycin (Invitrogen). The Jurkat and MS9 cells were supplemented with 10% or 20% heat-inactivated fetal calf serum (PAA Laboratories), respectively. Human embryonic kidney cells (HEK293T) were cultured in complete Dulbecco modified Eagle medium (Invitrogen) containing 50 IU/mL penicillin, 50 μg/mL streptomycin (Invitrogen), and 10% fetal calf serum.
Plasmids and transfection
Jurkat cells (clone E6.1) were transfected with 5 μg plasmid DNA using a Nucleofector device (Amaxa Biosystems) according to the manufacturer's optimized protocol (program X-05). Jurkat cells were transfected with the full-length tax gene in the pGFPC1 plasmid (Clontech),41 in which the gene is under the control of the CMV promoter. To express the fusion protein GFP-Tax, BamH1-EcoR1 fragment of Tax ORF was cloned into BglII-EcoR1 sites of pGFPC1. A series of 12 Tax mutants was generated by the substitution of amino acids in this GFP-Tax template. Tax mutants M7, M22, and M47, linked to specific functions during HTLV-1 infection, were generated by the substitution of 2 adjacent amino acid residues (Table 1). Tax ubiquitin mutants were generated by replacing lysine residues, known to be potential sites of ubiquitination, with arginine residues (Table 1).22,35,42,,,,,,,,,,–53
Preparation of Tax MVA
The Tax gene was cloned into modified vaccinia virus Ankara (MVA) as previously described.54 Recombinant virus was purified by 5 successive rounds of plaque purification. Bulk stocks were purified by centrifugation of cytoplasmic extracts of infected cells through a 36% (wt/vol) sucrose cushion in a Sorvall HB-4 rotor at 13 000g for 60 minutes.
Immobilization of antibodies on latex beads, conjugate formation, and analysis of MTOC polarization
The monoclonal antibodies anti–ICAM-1 and anti-CD3 (100 μg each) were adsorbed to 80 × 106 surfactant-free sulfate white latex 5-μm beads (Interfacial Dynamics Corporation), and cell-bead or cell-cell conjugates were allowed to form in 6-mm-diameter wells on a 10-well glass slide, as previously described.13,14 Cell conjugates were identified using a Leitz fluorescent microscope with a water-based objective (50×/1.0 NA). Between 50 and 100 events were counted per condition per experiment. A conjugate was scored only when the infected cell was in contact with a single uninfected cell (or single bead). We considered the MTOC of a donor cell to be polarized when it was located within one-fourth of the cell circumference oriented toward the contact with the target cell (or bead).
Indirect immunofluorescence staining for confocal microscopy
The cells were fixed either with paraformaldehyde 4%, for 20 minutes at room temperature, or with cold methanol, for 2 minutes at −20°C. After fixation, the cells were stained by immunofluorescence, as previously described.14 Confocal images were collected using Pascal LSM confocal imaging system (Carl Zeiss), equipped with a krypton-argon mixed-gas laser and a 63×/1.4 NA oil objective. Image processing was performed with the online LSM 5 Image Browser (Carl Zeiss). The images were analyzed with image analysis software (Scion image) and mounted with Photoshop 7.0 software.
Dual-luciferase assay
HEK293T cells were plated in Costar 24-well culture plate and cotransfected with NF-κB-luciferase55 or SRE-luciferase reporter plasmids, provided by Dr Melanie Cobb, and 100 ng/well of either wt pCMV4-Tax or the respective pCMV-Tax mutants M7, M22, M47, K280R, or K284R. Transfection with pRSV-Renilla served as the internal standard control. Cells were transfected with pCMV5-ΔMEKK1 and pCMV5-myc-RasV12 as positive controls for the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, respectively. Each experiment was carried out in triplicate using Fugene-6 (Roche Diagnostics) as transfection reagent. Cells were washed with 200 μL/well of phosphate-buffered saline and lysed with 100 μL/well of Passive Lysis Buffer (Promega). Cells were lysed 36 hours after transfection; the lysates were centrifuged at 13 000g for 2 minutes at 4°C. The luciferase assays were performed on FLUOstar Optima (BMG Labtech) using 20 μL of lysate per well in Costar 96-well plates as described by the manufacturer (Promega).
Statistical analysis
The frequency and statistical significance of MTOC polarization in HTLV-1–infected and uninfected cells were compared using the odds ratio as described previously.56 To calculate the summary odds ratio for a given effect over several experiments, we used the inverse variance method of weighting individual values of Ln(odds ratio).56 To calculate the statistical significance of a given effect over different experiments, we used the Fisher χ2 method of combining probabilities:
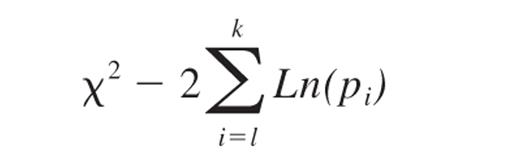
where pi is the significance level obtained from the ith experiment and k is the number of experiments; −2ΣLn(pi) is distributed as a χ2 variant with 2k degrees of freedom.57
Results
MTOC polarization in Jurkat cells transfected with wild-type or mutant Tax
We examined several Tax protein mutants (Table 1) for their ability to induce MTOC polarization in Jurkat T cells. The cells were transiently transfected with pGFPC1-Tax (wt) or with pGFPC1-Tax mutant; at 6 hours after transfection, the cells expressing Tax or its mutants were separately mixed with untransfected Jurkat cells (ratio 1:1) and then allowed to form cell-cell conjugates for 1 hour at 37°C. Using a conventional fluorescence microscope, we examined the conjugates formed between cells expressing GFP-Tax or its mutants (Tax+) and mock-transfected cells (Tax−). The Tax− or Tax mutant–positive cells in which the MTOC was oriented toward the Tax− cells were counted; the scores are reported in supplemental Tables 1 through 5 (available on the Blood website; see the Supplemental Materials link at the top of the online article). The results (Figure 1) showed that 90% of Jurkat cells expressing Tax protein (wt) had their MTOC oriented to the cell-cell contact region formed with untransfected Jurkat. By contrast, the cells expressing M7, M22, or M47 all failed to polarize their MTOC toward the target cell (Figure 1).

Frequency of MTOC polarization to the cell-cell junction induced by wild-type or mutant HTLV-1 Tax protein. Jurkat cells were transfected with expression plasmids encoding either wild-type or mutant Tax protein. After 6 hours, the cells were allowed to form conjugates for 1 hour at 37°C. In 2-cell conjugates (n = 50-100 in each experiment) formed between Tax+ and Tax− cells, we scored the frequency of polarization of MTOC in the Tax+ cell to the cell-cell junction. The bars represent the mean plus or minus SE of 3 to 5 independent experiments.
Frequency of MTOC polarization to the cell-cell junction induced by wild-type or mutant HTLV-1 Tax protein. Jurkat cells were transfected with expression plasmids encoding either wild-type or mutant Tax protein. After 6 hours, the cells were allowed to form conjugates for 1 hour at 37°C. In 2-cell conjugates (n = 50-100 in each experiment) formed between Tax+ and Tax− cells, we scored the frequency of polarization of MTOC in the Tax+ cell to the cell-cell junction. The bars represent the mean plus or minus SE of 3 to 5 independent experiments.
Tax ubiquitin mutant K280R failed to induce MTOC polarization
We investigated whether MTOC polarization induced by Tax protein is altered by mutation of the lysine residues that have been demonstrated to influence the ubiquitination of Tax.22,35 To prevent the addition of ubiquitin molecules to Tax protein, we made a series of mutations where the lysine residues were replaced with arginine residues. The results (Figure 1) show that mutation of Tax protein at position 280 (K280R) significantly diminished the frequency of MTOC polarization in the transfected donor T cells toward the untransfected recipient T cells (Figure 1). The mutations of Tax at either position 263 or 284 also caused a statistically significant but less marked reduction of the frequency of MTOC polarization (Figure 1; supplemental Tables 1–5). A double mutation of both the lysine at position 263 and 284 had no significant effect on Tax-induced MTOC polarization, but a triple mutation at positions 263 and 280 and 284 reduced considerably the frequency of this polarization (Figure 1). In contrast, mutation at position 189 or 193 or 197 in the N-terminal region of Tax protein had no effect. Point mutations of lysine at positions 85, 88, 111, 324, and 346 also had no effect on MTOC polarization induced by Tax (data not shown).
Inhibition of CREB activation abolished the MTOC polarization in Jurkat T cells infected with Tax recombinant MVA
Tax protein mutants M7 and M47, which failed to induce MTOC polarization in Jurkat cells (Figure 1), are both known to be deficient in activating the CREB signaling pathway. The M7 mutation prevents the interaction of Tax with the CREB protein,42 and the mutant M47 is deficient in CREB/activated transcription factor activation.43,44 To investigate whether inhibition of the CREB activity has a direct effect on the MTOC polarization, we used a selective inhibitor of Ca2+/calmodulin-dependent protein kinase kinase, STO-609, to block CREB activation.58 Jurkat cells were infected with the Tax recombinant MVA at a multiplicity of infection = 10 pfu/cell, incubated overnight at 37°C then treated with 10 μM of STO-609 for 2 hours at 37°C. After the treatment, the infected cells were mixed with uninfected and untreated Jurkat cells (ratio 1:1) and allowed to form conjugates for 1 hour at 37°C. The results (Figure 2) indicate that inhibition of CREB activation with STO-609 abolished the Tax-induced MTOC polarization in Jurkat cells.

Effect of inhibition of CREB phosphorylation on MTOC polarization in Jurkat cells expressing Tax protein. Jurkat cells were infected with the Tax recombinant MVA at a multiplicity of infection = 10 pfu/cell. After overnight Tax expression, the Jurkat T cells were treated (or mock-treated) with 10 μM of STO-609 for 1 hour at 37°C, then mixed with uninfected and untreated Jurkat cells at a ratio of 1:1, and incubated at 37°C for 1 hour to allow cell-cell conjugate formation. The cells were fixed, and Tax protein, the microtubules, and the nucleus were stained by immunofluorescence using anti-Tax monoclonal antibody (Lt-4), anti–β-tubulin-CY3, and 4,6-diamidino-2-phenylindole (DAPI), respectively. In 2-cell conjugates (n = 50-100 in each experiment) formed between Tax+ and Tax− cells, we scored the frequency of polarization of MTOC in the Tax+ cell to the cell-cell junction. The bars represent the mean plus or minus SE of 3 independent experiments.
Effect of inhibition of CREB phosphorylation on MTOC polarization in Jurkat cells expressing Tax protein. Jurkat cells were infected with the Tax recombinant MVA at a multiplicity of infection = 10 pfu/cell. After overnight Tax expression, the Jurkat T cells were treated (or mock-treated) with 10 μM of STO-609 for 1 hour at 37°C, then mixed with uninfected and untreated Jurkat cells at a ratio of 1:1, and incubated at 37°C for 1 hour to allow cell-cell conjugate formation. The cells were fixed, and Tax protein, the microtubules, and the nucleus were stained by immunofluorescence using anti-Tax monoclonal antibody (Lt-4), anti–β-tubulin-CY3, and 4,6-diamidino-2-phenylindole (DAPI), respectively. In 2-cell conjugates (n = 50-100 in each experiment) formed between Tax+ and Tax− cells, we scored the frequency of polarization of MTOC in the Tax+ cell to the cell-cell junction. The bars represent the mean plus or minus SE of 3 independent experiments.
Intracellular distribution of GFP-Tax or GFP-Tax mutants in Jurkat cells
To investigate whether MTOC polarization is associated with the presence of Tax at the MTOC region, we examined the intracellular distribution of Tax protein or its mutants by confocal microscopy in Jurkat cells expressing either GFP-Tax or GFP-Tax mutant (Figures 3–4). At 6 hours after transfection, the MTOC was stained with anti–γ-tubulin polyclonal antibody (Figures 3–4). We compared the distribution of Tax protein wild-type14 with that of mutants M7, M22, and M47 (Figure 3). The top panel shows GFP-Tax protein (in green) present mainly in the nucleus, with a significant fraction in the cytoplasm close to the MTOC. In CD4+ T cells naturally infected with HTLV-1, this fraction of Tax is not only associated with the MTOC but also with the cis-Golgi compartment.14 The mutant M47 (Figure 3) had a distribution similar to that of Tax (Figure 3). The mutant M22 was present both in the nucleus and the cytoplasm but failed to accumulate at the MTOC region (Figure 3). Finally, the mutant M7 was present exclusively in the cytoplasm (Figure 3) but failed to accumulate at the MTOC region.

Intracellular distribution of wild-type and mutants of HTLV-1–Tax protein in transiently transfected Jurkat cells. Jurkat cells were transfected with GFP-Tax or GFP-Tax mutants M7, M22, or M47. Jurkat cells were fixed with cold methanol (6 hours after transfection) and permeabilized. The MTOC was stained with polyclonal anti–γ-tubulin antibody (red). Each panel shows projected images (x, z) corresponding to GFP-Tax or GFP-Tax mutant (green), MTOC (red), and the superposition of the 2 colors (merge). Scale bar represents 10 μm.
Intracellular distribution of wild-type and mutants of HTLV-1–Tax protein in transiently transfected Jurkat cells. Jurkat cells were transfected with GFP-Tax or GFP-Tax mutants M7, M22, or M47. Jurkat cells were fixed with cold methanol (6 hours after transfection) and permeabilized. The MTOC was stained with polyclonal anti–γ-tubulin antibody (red). Each panel shows projected images (x, z) corresponding to GFP-Tax or GFP-Tax mutant (green), MTOC (red), and the superposition of the 2 colors (merge). Scale bar represents 10 μm.

Intracellular distribution of HTLV-1–Tax protein ubiquitin mutants in transiently transfected Jurkat cells. Jurkat cells are transfected with GFP-Tax or GFP-Tax-K263R, GFP-Tax-K280R, GFP-Tax-K284R, or GFP-Tax-K263R-K284R. The cells are fixed with cold methanol (6 hours after transfection) and permeabilized. The MTOC is stained with polyclonal antibody anti–γ-tubulin (red). Each panel shows projected images (x, z) corresponding to GFP-Tax or GFP-Tax mutant (green), MTOC (red), and the superposition of the 2 colors (merge). Scale bar represents 10 μm.
Intracellular distribution of HTLV-1–Tax protein ubiquitin mutants in transiently transfected Jurkat cells. Jurkat cells are transfected with GFP-Tax or GFP-Tax-K263R, GFP-Tax-K280R, GFP-Tax-K284R, or GFP-Tax-K263R-K284R. The cells are fixed with cold methanol (6 hours after transfection) and permeabilized. The MTOC is stained with polyclonal antibody anti–γ-tubulin (red). Each panel shows projected images (x, z) corresponding to GFP-Tax or GFP-Tax mutant (green), MTOC (red), and the superposition of the 2 colors (merge). Scale bar represents 10 μm.
We also examined the distribution pattern of the ubiquitin Tax mutants in Jurkat cells. Figure 4 shows the intracellular distribution patterns obtained either with Tax protein mutants K263R, K280R, K284R or K263R-K284R. All Tax ubiquitin mutants tested except K280R showed a distribution similar to that of wild-type Tax. The mutant K280R was expressed both in the nucleus and in the cytoplasm; but unlike the wild-type, this mutant did not accumulate at the MTOC region (Figure 4).
To verify whether the level of Tax expression influences its efficiency to induce MTOC polarization, we evaluated the level of expression of Tax and its mutants in transfected Jurkat cells (or HEK293T) by Western blot (supplemental Figure 1A-C) and by flow cytometry (supplemental Figure 1D). The results show that the level of Tax expression correlated with the transfection efficiency (ie, the frequency of cells expressing the protein) (supplemental Figure 1E,C: P = .002; r2 = 0.75) rather than the level of expression per cell. Because we counted the frequency of MTOC polarization only in Tax+ cells, our results were not affected by variation in transfection efficiency. Furthermore, comparison between the intensity of expression of the mutants M7 and M47 (supplemental Figure 1A-C) and the frequency of MTOC polarization (Figure 1) confirmed that the ability of Tax to induce MTOC polarization per cell is distinct from the total level of its expression.
NF-κB and MAP-kinase pathways activation were not required for the ability of Tax protein to induce MTOC polarization
We used an in vitro luciferase assay to test the ability of Tax (wt) or Tax mutant proteins (M7, M22, M47, K280R, and K284R) to activate either NF-κB or MAP-kinase signaling pathways. Each of these mutants induced MTOC polarization with significantly lower frequency than the wild-type Tax (Figure 1; supplemental Tables 1–5). As expected, the results (Figure 5A) confirmed that Tax (wt) efficiently activates the NF-κB pathway. In addition, Tax expression significantly increased the activity of the MAP-kinase pathway (Figure 5B). MAP-kinase activation induced by Tax expression was 4-fold superior to that obtained with an empty vector and 10% less than that obtained with the RasV12 plasmid used as a positive control. The results (Figure 5A) showed that not only M22 was deficient in NF-κB activation but also the mutants M7 and K284R. However, the mutants M47 and K280R were able to induce NF-κB activation. Finally, the mutants M22 and K280R activated the MAP-kinase pathway; by contrast, the mutants M7, M47, and K284R failed to activate this pathway. Comparison between the ability of Tax or its mutants to induce MTOC polarization and the intracellular localization of Tax (Table 2) suggests 2 conclusions. First, the accumulation of Tax at the MTOC is necessary but not sufficient for Tax-induced MTOC polarization. Second, activation of the NF-κB and MAP-kinase pathways is not required for Tax-induced polarization of the MTOC.

Effect of wild-type Tax and various Tax mutant proteins on NF-κB and MAPK signaling pathways. (A) HEK293T cells were transfected with 100 ng of wt pCMV4-Tax or pCMV-Tax mutants and NF-κB-luciferase reporter. Thirty-six hours after transfection, cells were harvested and lysed with Passive Lysis Buffer (Promega). Luciferase activity was measured on a FLUOstar Optima (BMG). pCMV5-ΔMEKK1 was used as a positive control in this assay. (B) The assay was similarly performed for the MAPK pathway using SRE-luciferase reporter. pCMV5-myc-RasV12 was used as the positive control. Error bars represent mean ± SE of triplicates; results shown are representative of 5 or 6 independent assays.
Effect of wild-type Tax and various Tax mutant proteins on NF-κB and MAPK signaling pathways. (A) HEK293T cells were transfected with 100 ng of wt pCMV4-Tax or pCMV-Tax mutants and NF-κB-luciferase reporter. Thirty-six hours after transfection, cells were harvested and lysed with Passive Lysis Buffer (Promega). Luciferase activity was measured on a FLUOstar Optima (BMG). pCMV5-ΔMEKK1 was used as a positive control in this assay. (B) The assay was similarly performed for the MAPK pathway using SRE-luciferase reporter. pCMV5-myc-RasV12 was used as the positive control. Error bars represent mean ± SE of triplicates; results shown are representative of 5 or 6 independent assays.
Blocking the MEK/ERK signaling pathway inhibited MTOC polarization induced by the cross-linking of ICAM-1 on the surface of Jurkat cells expressing Tax protein
Cross-linking of ICAM-1 on the surface of HTLV-1–infected cells is sufficient to induce efficient MTOC polarization.13,14 ICAM-1 engagement is thought to activate at least 2 signaling pathways, involving, respectively, RhoA and Ras GTPases.59 Our previous evidence using a specific inhibitor of RhoA (clostridial toxin C3) indicated that MTOC polarization induced by cross-linking of ICAM-1 is independent of the inactivation of Rho A.14 To investigate whether the Ras-MEK-ERK signaling route controls the MTOC polarization in Tax-expressing cells, we scored the frequency of MTOC polarization induced by the cross-linking of ICAM-1 molecules at the surface of Jurkat cells in the presence of each of 2 inhibitors of ERK phosphorylation, PD9805960,–62 and U012663,–65 (Figure 6).

Effect of blocking the MEK-ERK signaling pathway on MTOC polarization induced by the cross-linking of CD3 or ICAM-1 on the surface of Jurkat cells transiently transfected with Tax or CD4+ cells naturally infected with HTLV-1. The graphs represent the percentage of Jurkat cells expressing GFP-Tax protein or GFP protein alone (A), and naturally infected T cells (B) whose MTOC was oriented to the cell-bead contact region. Six hours after transfection with the respective plasmid, the cells were treated for 1 hour at 37°C either with cycloheximide (20 μM) or PD98059 (20 μM) or U0126 (100 μM), and then MTOC polarization was induced by cross-linking CD3 or ICAM-1 at the cell surface using latex beads coated with monoclonal antibodies directed against either CD3 or ICAM-1. Between 50 and 200 events were counted per condition, per experiment. The bars represent the mean ± SE of 3 independent experiments.
Effect of blocking the MEK-ERK signaling pathway on MTOC polarization induced by the cross-linking of CD3 or ICAM-1 on the surface of Jurkat cells transiently transfected with Tax or CD4+ cells naturally infected with HTLV-1. The graphs represent the percentage of Jurkat cells expressing GFP-Tax protein or GFP protein alone (A), and naturally infected T cells (B) whose MTOC was oriented to the cell-bead contact region. Six hours after transfection with the respective plasmid, the cells were treated for 1 hour at 37°C either with cycloheximide (20 μM) or PD98059 (20 μM) or U0126 (100 μM), and then MTOC polarization was induced by cross-linking CD3 or ICAM-1 at the cell surface using latex beads coated with monoclonal antibodies directed against either CD3 or ICAM-1. Between 50 and 200 events were counted per condition, per experiment. The bars represent the mean ± SE of 3 independent experiments.
The results (Figure 6A) show that nearly 80% of Jurkat cells expressing GFP-Tax oriented their MTOC to the cell-bead contact region, whereas only 15% of T cells expressing GFP had their MTOC polarized to the bead-contact region. By contrast, cross-linking of CD3 on the cell surface induced a higher percentage of MTOC polarization in both cells expressing GFP-Tax and cells expressing GFP vector alone (Figure 6A). The MTOC polarization induced by the cross-linking of ICAM-1 on the surface of Tax-expressing cells was blocked by the treatment with either cycloheximide or PD98059 or U0126. However, these treatments had no effect on the polarization induced by the cross-linking of CD3 (Figure 6A). We analyzed ERK phosphorylation in Jurkat cells by Western blot under the same conditions (supplemental Figure 2A-B). Our results showed that these inhibitors reduced ERK phosphorylation by 2- to 3-fold in cells stimulated with anti–ICAM-1 (lanes 10 and 11) compared with those stimulated with anti-CD3 (lanes 5 and 6). This result indicates that ICAM-1-mediated MTOC polarization is dependent on Ras-MEK-ERK activation, contrasting with that induced by T-cell receptor (TCR) engagement. We conclude that distinct signaling pathways are involved in MTOC polarization during the formation of the virological synapse compared with the immunologic synapse.
Inhibition of Ras-MEK/ERK signaling pathway abolishes MTOC polarization in naturally infected CD4+ and reduces HTLV-1 transfer between T cells
We investigated the effect of the inhibition of ERK phosphorylation on MTOC polarization in CD4+ T cells naturally infected with HTLV-1. These cells were isolated from the PBMCs of patients with HTLV-1–associated myelopathy/tropical spastic paraparesis, as previously described.13,14 The results (Figure 6B) show that the MTOC polarization was significantly reduced in the presence of ERK phosphorylation inhibitors only when the cells were stimulated by cross-linking of ICAM-1. This is consistent with the results obtained in Jurkat cells (Figure 6A) and supports the conclusion that MTOC polarization in HTLV-1–infected T cells depends on Ras-MEK/ERK signaling.
To confirm the physiologic relevance of this mechanism, we quantified the transfer of HTLV-1 proteins between MS9 cells (HTLV-1–producing cells) and uninfected Jurkat cells. The kinetics of HTLV-1 Gag transfer showed a peak intensity between 90 and 210 minutes after initiation of conjugate formation (supplemental Figure 3). This transfer was significantly reduced between 100 and 160 minutes either by depolymerization of the cytoskeleton (cytochalasin B, nocodazole), or by inhibition of ERK phosphorylation (PD98059, U0126), or by inhibition of CREB activation (STO-609). These kinetics (supplemental Figure 3) closely resembled the kinetics of the cell-cell transfer of HIV-1-Gag recently reported.66 These authors observed a peak intensity of HIV-Gag accumulation in target cells between 90 and 210 minutes of initiating cell-cell contact; HIV-1-Gag transfer was observed between 100 and 160 minutes. This time window (100-160 minutes) corresponds to the period during which HTLV-1–Gag transfer was most susceptible to inhibition with drugs. We hypothesize that this is the physiologically important window of HTLV-1 transfer between T cells.
Discussion
We previously showed that Tax protein expression is sufficient to sensitize the T cell to reorient its MTOC to the cell-cell contact region after engagement of ICAM-1 on the infected cell surface.13,14 We postulated that Tax protein expression and the engagement of ICAM-1 on the surface of the donor cell synergize to induce MTOC polarization. In the HTLV-1–infected T-lymphocyte (CD4+), Tax protein is located mainly at the nucleus but also accumulated closely in association with the MTOC region. Tax is also found in the cell-cell contact region formed between an infected donor cell and a target cell. A similar distribution of Tax protein was observed in Jurkat cells transiently transfected with pJFE-Tax or pGFPC1-Tax.14 The precise location of the MTOC fraction of Tax remains unclear. In naturally infected PBMCs, we previously showed that the fraction of Tax present at the MTOC is closely associated with the cis-Golgi compartment; we have confirmed this association by treating cells with either nocodazole or brefeldin A, which disperse Tax accumulated at the MTOC.14 This finding was recently corroborated by others.36,67 However, others have observed that Tax protein in HEK293 cells is present in a signalosome associated with the centrosome.68
In the present study, we found that introducing specific mutations in the Tax protein modified both its intracellular distribution and its ability to induce MTOC polarization. The mutants M7, M22, and K280R in particular showed a distribution different from that observed with the wild-type Tax protein. Unlike the wild-type, these mutants all failed to induce MTOC polarization toward the target cells or to accumulate at the MTOC region. These observations suggest an association between the presence of Tax protein at the MTOC region and its ability to induce MTOC polarization. However, this association did not apply in the case of the mutant M47, which failed to induce MTOC polarization even though its accumulation was clearly observed at the MTOC region. We conclude that the presence of Tax at the MTOC region is required but not sufficient to induce MTOC polarization in T cells expressing Tax.
Tax ubiquitination occurs at the carboxy-terminal half of Tax, especially at amino-acid positions 263, 280, and 284.35 It was reported that Tax protein ubiquitination is an important step in the Tax-induced activation of the NF-κB pathway.36,37 One of the mechanisms proposed suggests that Tax activates NF-κB by binding to IKK, in a centrosome-associated signalosome.68 The mutation of the lysine residues on Tax protein not only affects the posttranslational modifications of Tax, such as ubiquitination/sumoylation or acetylation, but also its intracellular distribution.36,37,69 Each of the lysine residues at positions 263, 280, and 284 contributes individually to Tax ubiquitination, whereas sumoylation requires the integrity of both lysine residues at positions 280 and 284.36,37 Tax ubiquitination is greatly reduced when the lysine residues in positions 263, 280, and 284 are mutated either individually or in combination. The reintroduction of lysine in position 280 alone partially restores Tax ubiquitination but does not restore NF-κB activity, whereas the reintroduction of lysine in position 284 has no effect.36,37 Our results indicate that, of these amino acid residues, only the lysine residue at position 280 is essential for the observed localization of Tax at the MTOC region, and also for the MTOC polarization caused by the cross-linking of ICAM-1 at the cell surface. The mutant K280R is able to activate efficiently the NF-κB signaling pathway. This is consistent with the results shown by others37,67 and suggests that NF-κB activation is not required for MTOC polarization. The failure of the mutant M47 to induce MTOC polarization despite its effective activation of NF-κB is also consistent with this conclusion. HTLV-1 Tax protein interacts directly with CREB to form a ternary complex on the HTLV-1 21-bp repeat; both the N terminus and C terminus of Tax are critical for Tax association and ternary complex formation.70 Tax and CREB together recruit CBP/p300 and coordinate the assembly of the transcriptional apparatus at the HTLV-1 promoter, resulting in high-level viral transcription.45,71,72 The mutants M7 (C29A, P30S) and M47 (L319R, L320S) are impaired in CREB activation,42 and we found that both failed to induce MTOC polarization. In addition, we showed that blocking the CREB activity with a CaMKK inhibitor (STO-609) abolished polarization of the MTOC in Tax-expressing cells. This drug did not perturb Tax association with the MTOC. These results corroborate the conclusion that CREB activation controls MTOC polarization and indicate that CREB activation by HTLV-1 Tax protein is essential for efficient MTOC polarization in HTLV-1–infected T cells.
We showed that Tax protein and the mutants M22 and K280R activated the MAPK signaling pathway in a cell-free assay, whereas the mutants M47, M7, and K284R did not (Table 2). This result suggests that activation of the MAPK pathway by Tax protein is not required for Tax-induced MTOC polarization because the mutant K284R induces a significant polarization of the MTOC. The mutant K284R retains the ability to activate CREB in vitro,37 further strengthening our conclusion that CREB activation is required for Tax-induced MTOC polarization.
We reported elsewhere that cross-linking of ICAM-1 at the cell surface induces MTOC polarization in HTLV-1–infected cells.13,14 It has been shown that the expression of HTLV-1 accessory protein p12 induces LFA-1 clustering on the surface of infected T cells via a calcium-dependent signaling pathway, which is thought to promote HTLV-1 spread between T cells.17 Elsewhere, p12 was reported to reduce the expression of ICAM-1/2 on the surface of CD4+ T cells, by an unknown mechanism; this reduction is thought to prevent the destruction of HTLV-1–infected cells by NK cells.18 However, HTLV-1 Tax expression was also shown to strongly up-regulate ICAM-1 expression73 : this effect may compensate for the effect of p12.18
ICAM-1–mediated signaling in T cells involves 2 pathways: one is RhoA-dependent,74 and the other involves Ras-MEK-ERK activation.75 We previously showed that MTOC polarization in HTLV-1–infected cells is independent of RhoA GTPase activation.14 Our present results confirm the involvement of the Ras-MEK-ERK pathway in MTOC polarization induced by the cross-linking of ICAM-1, both on the surface of T cells transfected with Tax and in T cells naturally infected with HTLV-1. Although we show that Tax-induced MTOC polarization is independent of MAPK activation, our results show that ICAM-1–induced MTOC polarization is dependent on the MEK/ERK pathway. Blocking the Ras-MEK-ERK pathway did not abolish MTOC polarization induced by cross-linking of CD3; nor did blocking protein synthesis using cycloheximide.
These results suggest that cross-linking of ICAM-1 at the cell surface activates a distinct pathway from that activated by TCR engagement, consistent with our previous observation that HTLV-1 infection significantly reduced the frequency of MTOC polarization caused by cross-linking TCR (CD3) on the cell surface,13 which suggested a competition between the 2 pathways. It is well established that TCR, LFA-1, and CD28 play distinct and complementary roles in causing T-cell cytoskeleton reorganization and MTOC reorientation.76 Costimulation through TCR/CD3 and accessory receptors (eg, CD28 or CD2) activates PKC, Ras, and Ras effectors MEK and PI3K. This activation induces the reorganization of the cytoskeleton at the contact region.77,78 Consistent with our present results, others have shown that blocking the MEK/ERK activation pathway in Jurkat cells with PD98059 had no effect on MTOC polarization induced by the cross-linking of the TCR.79 This suggests that MTOC reorientation induced by TCR signaling is not MEK/ERK-dependent. It was suggested that MTOC polarization in the T cell can occur via PI3K, another Ras effector molecule, which was shown to be required for TCR-stimulated ERK activation in cytotoxic T lymphocytes.80 Recent reports showed that CD28-stimulated MTOC polarization in YTS NK cells is dependent on the activation of ERK2.31 In addition, both ERK2 and JNK1 activation were shown to be essential for the polarization of both the MTOC and the cytotoxic granules in human NK cells.32 Therefore, it seems that simultaneous activation of 2 MAP-kinase pathways is required for polarization of the cytotoxic granules and MTOC during the formation of the cytotoxic T lymphocyte immunologic synapse.33
HTLV-1 Tax protein binds directly to IKK-γ, a part of the IKK complex, and this interaction correlates with Tax-induced IκB-α phosphorylation and NF-κB activation.81 Tax also binds directly to the protein kinase MEKK1, which contributes to Tax-mediated IKK activation in transfected cells.82 The interaction between MEKK1 and Tax might play a part in the synergistic induction of MTOC polarization by Tax and ICAM-1. However, Tax protein interacts with a large range of cellular proteins, including Ras p21 proactive 2 and cdc42/Rac effector kinase,83 which are a part of the Ras activation pathway.
Our results suggest a model in which the pathways involved in the MTOC polarization during the formation of the HTLV-1 virological synapse are distinct from those involved in MTOC polarization caused by the cross-linking of the TCR. Three factors are required for the synergistic polarization observed during the formation of the HTLV-1 virological synapse: (1) the presence of Tax protein in close association with the MTOC region in the infected T cell, (2) activation of the CREB pathway by Tax, and (3) activation of the MEK/ERK pathway by cross-linking of ICAM-1 at the cell surface. Finally, we found that the early cell-cell transfer of HTLV-1 was reduced by the inhibition of either Ras-MEK-ERK- or CREB-signaling pathways, consistent with the kinetics of HIV-1 transfer between T cells recently reported.66 We hypothesize that this is the physiologically important window of cell-cell transfer of HTLV-1 via the virological synapse.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dr Kuan-Teh Jeang for providing us with the pGFPC1-Tax construct, Dr Melanie Cobb for providing us with SRE-luciferase reporter, and Dr Aileen G. Rowan for helping with the statistical analysis.
This work was supported by the Wellcome Trust, United Kingdom.
Wellcome Trust
Authorship
Contribution: M.V.N. designed and performed the research, analyzed and interpreted the data, and wrote the manuscript; V.S.N. and S.M. performed experiments with luciferase reporter plasmids; Y.T. contributed by providing vital reagents (anti-Tax antibody); K.O. analyzed the data and contributed to the writing; G.P.T. contributed by providing vital blood samples; and C.R.M.B. designed the research, analyzed and interpreted the data, and contributed to the writing.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Charles R. M. Bangham, Department of Immunology, Wright-Fleming Institute, Imperial College, St Mary's Campus, Norfolk Pl, London W2 1PG London, United Kingdom; e-mail: c.bangham@imperial.ac.uk.

