

Key Points
Hemostatic plugs develop a regional architecture defined by the extent of platelet activation and packing density.
The regional composition of a hemostatic plug is due to the interaction of local conditions with the platelet-signaling network.
Abstract
Achieving hemostasis following vascular injury requires the rapid accumulation of platelets and fibrin. Here we used a combination of confocal intravital imaging, genetically engineered mice, and antiplatelet agents to determine how variations in the extent of platelet activation following vascular injury arise from the integration of different elements of the platelet-signaling network. Two forms of penetrating injury were used to evoke the hemostatic response. Both produced a hierarchically organized structure in which a core of fully activated platelets was overlaid with an unstable shell of less-activated platelets. This structure emerged as hemostasis was achieved and persisted for at least 60 minutes following injury, its organization at least partly reflecting agonist concentration gradients. Thrombin activity and fibrin formation were found primarily in the innermost core. As proposed previously, greater packing density in the core facilitated contact-dependent signaling and limited entry of plasma-borne molecules visualized with fluorophores coupled to dextran and albumin. Blocking contact-dependent signaling or inhibiting thrombin reduced the size of the core, while the shell was heavily influenced by adenosine 5′-diphosphate and regulators of Gi2-mediated signaling. Thus, the hemostatic response is shown to produce a hierarchical structure arising, in part, from distinct elements of the platelet-signaling network.
Introduction
Platelet accumulation is a hallmark of hemostasis and a contributing factor in heart attacks and strokes. Platelet activation is driven by receptor-mediated signaling in response to stimuli of varying potency, such as collagen, thrombin, adenosine 5′-diphosphate (ADP), and thromboxane A2 (TxA2). This has led to a model of the hemostatic response in which redundant elements of the platelet-signaling network work in concert to produce platelet aggregation, thrombin generation, and a hemostatic mass composed of activated platelets interspersed with fibrin. Interestingly, despite the long-recognized ability of multiple platelet agonists to drive platelet activation to completion in vitro, observations performed in vivo show that platelet activation is not uniform throughout a hemostatic plug. Rather, some of the platelets accumulating at a site of injury retain a discoid, or “resting,” morphology,1-3 cytosolic calcium mobilization is heterogeneous,4,5 and α-granule secretion occurs nonuniformly throughout the growing hemostatic mass.6-8 Consistent with these recent observations performed in vivo, variations in the extent of platelet activation during the hemostatic response have been demonstrated by electron microscopy studies dating back to the 1960s that examined thrombi formed in vivo and ex vivo.9-11
These observations raise a number of questions. If the hemostatic response normally produces a mixed population of platelets with varying degrees of activation, what are the implications for achieving a stable plug and for avoiding unnecessary vascular occlusion? How can a common signaling network produce distinguishable outcomes among participating platelets and how might different agonists contribute to these outcomes? How does the growing hemostatic structure alter the conditions experienced by individual platelets and what impact does that have on subsequent events? Finally, how might differences in the clinical impact of antiplatelet agents taken to prevent adverse cardiovascular events be understood in the context of the heterogeneous platelet activation observed during the hemostatic response?
With these questions in mind, our first goal in the present study was to determine how variations in platelet activation in vivo arise through the integration of distinct elements of the platelet-signaling network. Our second goal was to determine how regional variations in the extent of platelet activation affect the stability of the hemostatic mass and the passage of plasma-borne molecules within the mass. To achieve these goals, we used a combination of high-resolution intravital confocal microscopy, genetically engineered mice, and well-characterized antiplatelet agents to examine the hemostatic response produced by 2 types of penetrating injury. In the first, a laser was used to make a defect large enough to allow red cells as well as plasma to escape. In the second, a sharpened glass micropipette was used to produce a penetrating injury without the heat produced by the laser.
The results in both cases show that the hemostatic response produces a hierarchical structure in which a core of closely packed, irreversibly activated platelets is overlaid by a shell of loosely associated, minimally activated platelets. In addition, using fluorescent markers as probes, we showed that close platelet packing within the core reduces plasma volume in this region, increases resistance to the penetration of large plasma-borne molecules, and enables contact-dependent signaling. The region of the core most proximal to the vascular wall is the primary site of thrombin activity and fibrin deposition. However, reflecting the penetrating nature of the injury, fibrin formation also extends into the extravascular space. As suggested by earlier studies,5 inhibition of thrombin activity significantly impairs full platelet activation and formation of a definable core region. In the present study we also show that contact-dependent signaling by semaphorin 4D12,13 regulates the size of the core, while shell size is most heavily influenced by ADP and regulators of Gi2-mediated signaling. Thus, the ability of multiple agonists to activate platelets in vitro can be seen not as representing redundancy but as a means to sculpt and limit the hemostatic response in vivo.
Methods
Mice
C57Bl/6J mice (Jackson Laboratories, Bar Harbor, ME) were used for all studies examining wild-type mice. Semaphorin 4D–deficient mice were described previously.14 They are congenic on the C57Bl/6 background, although studies examining hemostatic plug formation in these mice compared wild-type (Sema4D+/+) and sema4D deficient (Sema4D−/−) littermates produced from crosses of heterozygous (Sema4D+/−) mice. Gi2α(G184S) mutant mice were provided by Dr. Richard Neubig (University of Michigan) and have been described previously.15 Mice used in this study were heterozygous for the mutant allele (Gnai2+/G184S) or wild-type (Gnai2+/+) littermates. The University of Pennsylvania Institutional Animal Care and Use Committee approved all procedures.
Vascular injury in the mouse cremaster muscle microcirculation
Hemostatic plug formation was visualized in the cremaster muscle microcirculation of male mice according to a modification of procedures originally developed by Falati et al.16 A detailed description of the equipment, procedures, antibodies/fluorophores, and injury models used is included in the supplemental Materials.
Hemostatic plug porosity calculations
Hemostatic plug porosity was calculated using the fluorescence intensity of plasma phase tracer molecules (dextrans or albumin) infused intravenously during intravital microscopy experiments. Fluorescently tagged albumin or dextrans of various molecular weights were infused via the jugular vein cannula 45 to 90 seconds post-injury and again 20 minutes post-injury to illuminate the plasma (50 µg per infusion). An optical plane through the center of the platelet mass was imaged by spinning disk confocal microscopy. The tracer, anti-CD41, and anti-P-selectin channel fluorescence were acquired using time-lapse imaging and saved for subsequent offline analysis. Hemostatic plug porosity was calculated using the tracer fluorescence intensity within the platelet mass compared with the tracer intensity in the whole blood (ie, in the vessel lumen) according to the following equation:
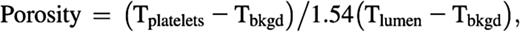
where Tplatelets is the mean tracer fluorescence intensity within the platelet mass (or a subregion of the platelet mass), Tbkgd is the mean background tracer intensity measured in the extravascular space, and Tlumen is the mean tracer intensity within the blood vessel lumen. The Tlumen is multiplied by 1.54 to account for the volume of blood occupied by red blood cells (this correction factor was determined empirically). The total hemostatic plug area was defined by the anti-CD41 fluorescence, the area of the core region was defined by P-selectin fluorescence, and the CD41-positive–area minus the P-selectin–positive area defined the outer shell region.
Statistical analysis
All data were analyzed using GraphPad Prism 5.0 and are presented as means ± standard error of the mean (SEM). Statistical analysis was performed using a 2-tailed student’s t test or 2-way analysis of variance (ANOVA) with the Bonferroni post-test for multiple groups as indicated in the legends of Figures 4–F5,6.
Results
Regional differences in platelet activation within the hemostatic plug
We began by examining hemostasis following penetrating injuries to cremaster muscle arterioles in adult C57Bl/6 mice. P-selectin surface expression was used as a marker for α-granule secretion and irreversible platelet activation. Injuries were created with either a laser or a glass micropipette. Loss of blood prior to hemostasis was demonstrated by the extravasation of red cells and fluorescently labeled albumin (supplemental Video 1). Blood loss ended as soon as small numbers of platelets accumulated, after which extravascular albumin diffused rapidly away from the injured vessel. However, escape of albumin and red cells continued when similar injuries were performed in mice rendered thrombocytopenic by injection of an anti-GPIb antibody (data not shown). Thus, both laser and micropuncture injuries may be considered models of hemostasis.
The hemostatic plugs that formed following either laser or micropuncture injury were qualitatively similar. In both cases, we observed an initial growth phase corresponding to net platelet accumulation, followed by a phase of net platelet loss (Figure 1A and supplemental Video 1). As others have noted, surface expression of P-selectin following injury in arterioles was delayed relative to platelet accumulation6,7 and, in contrast to venules (where endothelial cells also bound the antibody), was limited to platelets (Figure 1 and supplemental Figure 1). It initially occurred immediately adjacent to or within the vessel wall at the site of injury, regardless of the form of injury (Figure 1A–C). The number of P-selectin–positive platelets increased over time but remained spatially restricted. When it occurred, embolization was limited to the P-selectin–negative platelet population (Figure 1D and supplemental Figure 2).

Heterogeneity of platelet activation following laser or micropuncture injury. (A) Graphs show total platelet accumulation (red line) and P-selectin expression (green line) following laser (upper) or micropuncture (lower) injury in mouse cremaster muscle arterioles. Data are expressed as mean area ± SEM. N = 21 laser injuries in 6 mice and N = 29 pipette injuries in 7 mice. (B) Two-dimensional confocal images 3 and 20 minutes after laser (upper panels) or micropuncture (lower panels) injury. Platelets are labeled with Alexa-fluor 568 anti-CD41 F(ab)2 fragments (red). Alexa-fluor 647 anti-P-selectin was used to label degranulated platelets (green). Yellow is the merge. (C) Images are 3-dimensional reconstructions (isosurface view) of a series of z-plane images from the same injuries shown in Figure 1B taken 5 minutes post-laser (Ci) or puncture (Cii) injury. P-selectin is shown in green and platelets (CD41) in red. Grid size is 15 µm. Time-lapse videos of the formation and 3D reconstructions of the hemostatic plugs in A–C are provided in supplemental Video 1. (D) P-selectin–positive platelets are stably adherent during hemostatic plug formation in vivo. Embolization area was calculated by summing the total area of CD41 or P-selectin–positive events occurring in a region downstream from the main platelet mass (see supplemental Figure 2 and Supplemental Methods for details). Values are the mean ± SEM of the total CD41 or P-selectin embolus area in the first 250 seconds following injury from n = 5 injuries for pipette injury and n = 5 injuries for laser injury.
Heterogeneity of platelet activation following laser or micropuncture injury. (A) Graphs show total platelet accumulation (red line) and P-selectin expression (green line) following laser (upper) or micropuncture (lower) injury in mouse cremaster muscle arterioles. Data are expressed as mean area ± SEM. N = 21 laser injuries in 6 mice and N = 29 pipette injuries in 7 mice. (B) Two-dimensional confocal images 3 and 20 minutes after laser (upper panels) or micropuncture (lower panels) injury. Platelets are labeled with Alexa-fluor 568 anti-CD41 F(ab)2 fragments (red). Alexa-fluor 647 anti-P-selectin was used to label degranulated platelets (green). Yellow is the merge. (C) Images are 3-dimensional reconstructions (isosurface view) of a series of z-plane images from the same injuries shown in Figure 1B taken 5 minutes post-laser (Ci) or puncture (Cii) injury. P-selectin is shown in green and platelets (CD41) in red. Grid size is 15 µm. Time-lapse videos of the formation and 3D reconstructions of the hemostatic plugs in A–C are provided in supplemental Video 1. (D) P-selectin–positive platelets are stably adherent during hemostatic plug formation in vivo. Embolization area was calculated by summing the total area of CD41 or P-selectin–positive events occurring in a region downstream from the main platelet mass (see supplemental Figure 2 and Supplemental Methods for details). Values are the mean ± SEM of the total CD41 or P-selectin embolus area in the first 250 seconds following injury from n = 5 injuries for pipette injury and n = 5 injuries for laser injury.

Fibrin localization partially overlaps with α-granule release in time and space. (A) Photomicrographs showing representative 2-dimensional confocal images illustrating localization of P-selectin and fibrin in a hemostatic plug following laser-induced injury. The image on the left shows the anti-fibrin (green) alone on the brightfield background. The image on the right shows platelets (CD41, red), P-selectin (blue), and fibrin (green) on the brightfield background; see color key at right for overlapping fluorophore colors. All 3 fluorescent channels are shown as binary images. Fibrin was imaged using an anti-fibrin antibody. (B) Representative photomicrographs illustrating localization of fibrin in the extravascular region adjacent to the site of laser injury. The image on the left shows Alexa-488–labeled fibrinogen on the brightfield background. The image on the right shows platelets (CD41, red), P-selectin (blue), and Alexa-488 fibrinogen (green) on the brightfield background. CD41 and P-selectin channels are displayed as binary images. The γ setting of the fibrinogen channel was set to 0.5 to increase the brightness of the extravascular fibrin network. Fibrin(ogen) (green) is observed throughout the vessel lumen and hemostatic plug, but discernible fibrin fibers are only observed in the extravascular space. Bars represent 10 µm. Time-lapse videos of the formation and 3-dimensional reconstructions of the hemostatic plugs in both A and B are provided in supplemental Video 2.
Fibrin localization partially overlaps with α-granule release in time and space. (A) Photomicrographs showing representative 2-dimensional confocal images illustrating localization of P-selectin and fibrin in a hemostatic plug following laser-induced injury. The image on the left shows the anti-fibrin (green) alone on the brightfield background. The image on the right shows platelets (CD41, red), P-selectin (blue), and fibrin (green) on the brightfield background; see color key at right for overlapping fluorophore colors. All 3 fluorescent channels are shown as binary images. Fibrin was imaged using an anti-fibrin antibody. (B) Representative photomicrographs illustrating localization of fibrin in the extravascular region adjacent to the site of laser injury. The image on the left shows Alexa-488–labeled fibrinogen on the brightfield background. The image on the right shows platelets (CD41, red), P-selectin (blue), and Alexa-488 fibrinogen (green) on the brightfield background. CD41 and P-selectin channels are displayed as binary images. The γ setting of the fibrinogen channel was set to 0.5 to increase the brightness of the extravascular fibrin network. Fibrin(ogen) (green) is observed throughout the vessel lumen and hemostatic plug, but discernible fibrin fibers are only observed in the extravascular space. Bars represent 10 µm. Time-lapse videos of the formation and 3-dimensional reconstructions of the hemostatic plugs in both A and B are provided in supplemental Video 2.
The resulting hemostatic plug was thus composed of at least 2 distinct populations of platelets: a core of stably adherent degranulated platelets and a loose shell of P-selectin–negative platelets that overlaid the core and formed a downstream tail (Figure 1B–C). Although the extent of total platelet accumulation was greater after laser injury than after puncture injury (Figure 1A), the regional features were the same, indicating that the core and shell are common features of the hemostatic response in this part of the circulation. Subsequent studies were performed using the laser injury model.
Endovascular fibrin deposition and formation of an extravascular fibrin network
Using an antifibrin antibody that does not recognize fibrinogen,17 we found that endovascular fibrin formation displayed similar kinetics to P-selectin expression (data not shown). Spatially, the fibrin partly overlapped with the P-selectin–positive platelets but also extended into the vessel wall (Figure 2A). Fibrin was not detectable in the outer shell of P-selectin–negative platelets or even in the outermost regions of the core. The endovascular distribution of fibrin is similar to the distribution of thrombin activity that we have recently observed with a novel thrombin sensor that binds to the platelet surface.18 For comparison, mice were infused with fluorescently labeled fibrinogen. Extravasation of the fibrinogen at the site of injury was followed by its rapid conversion into a fibrous network that remained in place (Figure 2B, supplemental Video 2); this is in contrast to fluorescently tagged albumin that rapidly diffused away (supplemental Video 1).
Packing density
Fully activated platelets are held close together by integrin αIIbβ3, the platelet fibrinogen receptor, producing a maze of narrow gaps and contacts between adjacent platelets. We have previously proposed that this would affect the influx of plasma-borne molecules to the inside of a platelet mass.19 We tested this hypothesis by comparing the ability of fluorescent tracers to penetrate the core and shell regions of hemostatic plugs, starting with fluorescently tagged anti-P selectin. As noted, when infused prior to injury, this antibody labels platelets throughout the core region (Figure 3A). However, when infused 25 minutes after injury, antibody binding was limited to platelets at the apparent boundary between the shell and core and did not include platelets beneath this boundary (Figure 3B). The P-selectin antibody used in these studies is an intact immunoglobulin G and specific for a secretion-dependent marker; however, identical results were obtained with anti-CD41 F(ab)2 fragments that bind to αIIbβ3 regardless of its activation state (supplemental Figure 3).

Plasma protein accessibility to the core region is limited. Photomicrographs showing representative hemostatic plugs from 2 different mice. (A) The anti-P-selectin antibody was infused prior to injury and circulating throughout hemostatic plug formation. (B) The anti-P-selectin antibody was infused 25 minutes post-injury. For both A and B, the platelet (i) and P-selectin (ii) channels are shown individually as gray-scale images and the merged image (iii) is shown in color (red, platelets; green, P-selectin). Bars represent 10 µm. A line drawing of the merged images is included for clarity (iv). Note that the P-selectin antibody is unable to penetrate the core region when infused 25 minutes post-injury.
Plasma protein accessibility to the core region is limited. Photomicrographs showing representative hemostatic plugs from 2 different mice. (A) The anti-P-selectin antibody was infused prior to injury and circulating throughout hemostatic plug formation. (B) The anti-P-selectin antibody was infused 25 minutes post-injury. For both A and B, the platelet (i) and P-selectin (ii) channels are shown individually as gray-scale images and the merged image (iii) is shown in color (red, platelets; green, P-selectin). Bars represent 10 µm. A line drawing of the merged images is included for clarity (iv). Note that the P-selectin antibody is unable to penetrate the core region when infused 25 minutes post-injury.
As a comparison with antibodies that bind to the platelet surface, we also measured the penetration of fluorescently labeled albumin and dextran. When albumin was infused >20 minutes post-injury, it filled the plasma volume within the platelet mass, illuminating the gaps between adjacent platelets (Figure 4A, supplemental Video 3). The plasma volume within hemostatic plugs was estimated based on its fluorescence intensity compared with the bulk blood flow and is reported as the porosity (see Methods for details). Calculated porosity was significantly less in the core region than in the shell (Figure 4B). Together with the antibody penetration studies, these results show that regions within the growing hemostatic plug can be distinguished by their packing density as well as their state of activation.

Access of plasma tracers to the core region is size dependent. (A) Representative photomicrographs showing Alexa-488–labeled albumin infused 25 minutes post-injury. The image at the left shows albumin fluorescence. The brightfield image on the right is overlaid with a pseudocolor image depicting albumin intensity within the hemostatic plug. The black line in the pseudocolor image outlines the P-selectin–positive area. A time-lapse video illustrating albumin penetration into this hemostatic plug is included in supplemental Video 3. (B) The graph shows the porosity measured using fluorescent albumin as a tracer for the shell and core regions. Values are mean ± SEM for n = 8 injuries from 4 mice; statistics were performed using a 2-tailed Student t test. (C) Photomicrographs showing representative hemostatic plugs 20 minutes post-injury following infusion of either 10-kDa dextran (Ci) or 70-kDa dextran (Cii). Images are pseudocolored to depict dextran concentration within the platelet mass (the pseudocolored region depicts the entire platelet mass as defined by CD41 fluorescence; blue corresponds to low dextran, red to high dextran). Note the lack of 70-kDa dextran penetration into the core (dark blue region in Cii). (D) The graph shows platelet mass porosity 20 minutes post-injury calculated using 3 different sized dextrans (3 kDa, open bars; 10 kDa, gray bars; and 70 kDa, black bars). Data are expressed as the mean ± SEM porosity within the core and outer shell regions. N = 13 injuries from 4 mice for the 3-kDa dextran, 8 injuries from 4 mice for the 10-kDa dextran, and 8 injuries from 4 mice for the 70-kDa dextran. Statistical comparisons made using a 2-way ANOVA with Bonferroni post-hoc test. NS indicates not significant. #, indicates apparent porosity because the true porosity should be probe-insensitive and the reduced value obtained with 70-kDa dextran represents inadequate access to the core region due to a transport barrier.
Access of plasma tracers to the core region is size dependent. (A) Representative photomicrographs showing Alexa-488–labeled albumin infused 25 minutes post-injury. The image at the left shows albumin fluorescence. The brightfield image on the right is overlaid with a pseudocolor image depicting albumin intensity within the hemostatic plug. The black line in the pseudocolor image outlines the P-selectin–positive area. A time-lapse video illustrating albumin penetration into this hemostatic plug is included in supplemental Video 3. (B) The graph shows the porosity measured using fluorescent albumin as a tracer for the shell and core regions. Values are mean ± SEM for n = 8 injuries from 4 mice; statistics were performed using a 2-tailed Student t test. (C) Photomicrographs showing representative hemostatic plugs 20 minutes post-injury following infusion of either 10-kDa dextran (Ci) or 70-kDa dextran (Cii). Images are pseudocolored to depict dextran concentration within the platelet mass (the pseudocolored region depicts the entire platelet mass as defined by CD41 fluorescence; blue corresponds to low dextran, red to high dextran). Note the lack of 70-kDa dextran penetration into the core (dark blue region in Cii). (D) The graph shows platelet mass porosity 20 minutes post-injury calculated using 3 different sized dextrans (3 kDa, open bars; 10 kDa, gray bars; and 70 kDa, black bars). Data are expressed as the mean ± SEM porosity within the core and outer shell regions. N = 13 injuries from 4 mice for the 3-kDa dextran, 8 injuries from 4 mice for the 10-kDa dextran, and 8 injuries from 4 mice for the 70-kDa dextran. Statistical comparisons made using a 2-way ANOVA with Bonferroni post-hoc test. NS indicates not significant. #, indicates apparent porosity because the true porosity should be probe-insensitive and the reduced value obtained with 70-kDa dextran represents inadequate access to the core region due to a transport barrier.
Accessibility of plasma-borne molecules to the core region is likely to be dependent on a number of factors, including hydrodynamic radius (Rh). To test this hypothesis, we calculated apparent porosity using fluorescent dextran molecules. Dextran was selected because it is biologically inert, has been used previously as a contrast agent in vivo,20 and is available in different sizes. Twenty minutes after injury we obtained similar estimates of porosity using either 3 kDa (Rh ∼1.5 nm) or 10 kDa (Rh ∼2.7 nm21 ) dextran (Figure 4C–D). Both tracers showed a significant difference between the shell and core (P < .01 for 3-kDa dextran), with porosity values similar to those obtained with fluorescently labeled albumin (Rh ∼3.5 nm). In contrast, 70-kDa dextran (Rh ∼6.4 nm21 ) penetrated the core less freely, resulting in a calculated porosity that was one-third less than 3-kDa dextran (Figure 4C–D, P < .05, supplemental Video 4). This decrease was limited to the core; calculated porosity in the shell was not significantly different among the dextrans tested (Figure 4D).
These results indicate that the gaps between platelets in the outer shell are large enough to allow equal access to all the tracers tested, while the greater packing density in the core region is sufficient to reduce entry of the largest tracer. Note that a core porosity of ∼0.4 indicates that 60% of the core volume is platelets; this represents a >120-fold increase in platelet concentration relative to mouse platelet–rich plasma where platelets occupy ∼0.5% by volume (5 fL mean platelet volume × 106 platelets/μL plasma).
Mapping the platelet signaling network to the core and shell
One goal in this study was to determine how different elements of the platelet-signaling network contribute to the organizational structure of the hemostatic response. We began by testing mice that either lack semaphorin 4D (Sema4D)13 or express the Gi2 gain of function mutation [Gi2α(G184S)].22 Sema4D is a cell surface molecule that participates in contact-dependent signaling by acting as a ligand for receptors on the surface of an adjacent platelet. We have shown previously that this reinforces platelet activation by collagen and that loss of Sema4D expression reduces platelet accumulation on collagen under flow when studied in vitro under conditions that permit platelet–platelet interactions.12,13 Because greater packing density would be expected to facilitate contact-dependent signaling, we hypothesized that loss of Sema4D would preferentially impact the core region, with only a secondary effect on formation of the outer shell. Consistent with our previous report,23 overall platelet accumulation measured with anti-CD41 was substantially reduced in Sema4D−/− mice (Figure 5A). Addition of anti-P–selectin antibodies now shows that this decrease is primarily due to a >50% reduction in the size of the core region; shell size was not significantly reduced (Figure 5A–B).

Platelet signaling mutants differentially impact the hemostatic plug core and shell. (A) Platelet accumulation (CD41) and P-selectin expression over time following laser injury in wild-type (WT, gray lines) and semaphorin 4D-deficient (Sema4D, black lines) mice. Values are mean area ± SEM for n = 33 injuries in 5 WT mice and n = 44 injuries in 7 Sema4D mice. (B) A comparison of shell and core area 2 minutes post-injury. Data are normalized to mean area of the wild-type for each region and are expressed as mean ± SEM. Statistical analysis was performed by 2-way ANOVA with Bonferroni post-hoc test. The 95% confidence intervals for the difference between means (µWT − µSema4D) are −0.6494 to 0.2299 and −0.9920 to −0.1127 for the shell and core, respectively. (C) Platelet accumulation (CD41) and P-selectin expression following laser injury in cremaster muscle arterioles of wild-type (WT, gray lines) and Gi2α(G184S) mice (black lines). Values are mean area ± SEM for n = 47 injuries in 6 WT mice and n = 48 injuries in 5 Gi2α(G184S) mice. (D) A comparison of shell and core area 2 minutes post-injury as in B.
Platelet signaling mutants differentially impact the hemostatic plug core and shell. (A) Platelet accumulation (CD41) and P-selectin expression over time following laser injury in wild-type (WT, gray lines) and semaphorin 4D-deficient (Sema4D, black lines) mice. Values are mean area ± SEM for n = 33 injuries in 5 WT mice and n = 44 injuries in 7 Sema4D mice. (B) A comparison of shell and core area 2 minutes post-injury. Data are normalized to mean area of the wild-type for each region and are expressed as mean ± SEM. Statistical analysis was performed by 2-way ANOVA with Bonferroni post-hoc test. The 95% confidence intervals for the difference between means (µWT − µSema4D) are −0.6494 to 0.2299 and −0.9920 to −0.1127 for the shell and core, respectively. (C) Platelet accumulation (CD41) and P-selectin expression following laser injury in cremaster muscle arterioles of wild-type (WT, gray lines) and Gi2α(G184S) mice (black lines). Values are mean area ± SEM for n = 47 injuries in 6 WT mice and n = 48 injuries in 5 Gi2α(G184S) mice. (D) A comparison of shell and core area 2 minutes post-injury as in B.
In contrast to the Sema4D knockout, platelets from Gi2α(+/G184S) mice display a gain of function phenotype because the substitution renders Gi2α insensitive to feedback inactivation by RGS proteins.15,22 This increases platelet function in vitro, in part, by increasing the duration of signaling downstream of the platelet ADP receptor, P2Y12, and enhances overall thrombus size in vivo.22 New measurements performed here show that this increase is due solely to expansion of the P-selectin–negative shell region (Figure 5C–D). Shell size reflects the net of platelet adhesion, platelet embolization, and conversion of platelets to a fully activated (ie, P-selectin–positive) state. Because core size was unaffected, these results suggest that sustained Gi2 signaling increases the likelihood that new platelets will adhere and remain in place without affecting their conversion to the fully activated state.
The impact of a thrombin inhibitor and a P2Y12 antagonist on hemostatic plug architecture
Two well-characterized inhibitors of thrombin and P2Y12 receptors were used to map the effects of thrombin and ADP on the hierarchical organization of the hemostatic response. Gi2 is the primary mediator of platelet responses downstream of the ADP receptor, P2Y12.24,25 Consistent with the results in the Gi2α(+/G184S) mice, addition of the direct-acting P2Y12 antagonist, cangrelor, significantly reduced the size of the hemostatic plug shell without substantially affecting the core region (Figure 6A–B and supplemental Video 5), indicating that ADP is critical for recruitment and retention of platelets in the shell. Thrombin, on the other hand, appears to be the primary factor in platelet activation within the core. Consistent with earlier reports,7,16 we found that a direct thrombin inhibitor, hirudin, substantially reduced both platelet accumulation and fibrin formation (Figure 6C–F and supplemental Video 6). Notably, very few of the platelets that adhered at the site of injury in the presence of hirudin became P-selectin positive. Thus, loss of thrombin activity greatly inhibits formation of the core region, leaving behind only a remnant of the unstable shell. Taken together, these results suggest that full platelet activation and core formation are driven by a gradient of thrombin emanating from the base of the hemostatic plug and site of injury, with region-specific reinforcement from ADP and contact-dependent signaling mechanisms in the outer shell and innermost core, respectively.

Contribution of ADP and thrombin to the formation of the hemostatic plug core and shell. (A) Graph depicts total platelet accumulation and P-selectin expression over time following laser injury in wild-type mice before (blue lines) and after (red lines) treatment with the P2Y12 antagonist cangrelor. Values are mean area ± SEM for n = 15 injuries before and 24 injuries after cangrelor infusion in 4 wild-type mice. Representative time-lapse videos are included in supplemental Video 5. (B) A comparison of shell and core area 2 minutes post-injury. Data are normalized to mean area of the wild-type for each region and are expressed as mean ± SEM. Statistical analysis was performed by 2-way ANOVA with Bonferroni post-hoc test. (C–F) Impact of thrombin inhibition on the core and shell. Representative photomicrographs show hemostatic plugs 2 minutes post-injury before (C) and after (D) intravenous infusion of hirudin (30 µg/kg prior to injury). Platelets are labeled red, P-selectin is labeled green, and fibrin is labeled blue. Overlay of platelets and P-selectin is yellow and overlay of all 3 fluorophores is white. (E) and (F) Kinetics of hemostatic plug formation for the 2 injuries shown in (C) and (D), respectively. The red line is total platelet accumulation (CD41 area), the green line is P-selectin expression, and the blue line is fibrin accumulation. Time-lapse videos of the hemostatic plugs shown are included in supplemental Video 6. Data are representative of at least 6 injuries in 2 mice.
Contribution of ADP and thrombin to the formation of the hemostatic plug core and shell. (A) Graph depicts total platelet accumulation and P-selectin expression over time following laser injury in wild-type mice before (blue lines) and after (red lines) treatment with the P2Y12 antagonist cangrelor. Values are mean area ± SEM for n = 15 injuries before and 24 injuries after cangrelor infusion in 4 wild-type mice. Representative time-lapse videos are included in supplemental Video 5. (B) A comparison of shell and core area 2 minutes post-injury. Data are normalized to mean area of the wild-type for each region and are expressed as mean ± SEM. Statistical analysis was performed by 2-way ANOVA with Bonferroni post-hoc test. (C–F) Impact of thrombin inhibition on the core and shell. Representative photomicrographs show hemostatic plugs 2 minutes post-injury before (C) and after (D) intravenous infusion of hirudin (30 µg/kg prior to injury). Platelets are labeled red, P-selectin is labeled green, and fibrin is labeled blue. Overlay of platelets and P-selectin is yellow and overlay of all 3 fluorophores is white. (E) and (F) Kinetics of hemostatic plug formation for the 2 injuries shown in (C) and (D), respectively. The red line is total platelet accumulation (CD41 area), the green line is P-selectin expression, and the blue line is fibrin accumulation. Time-lapse videos of the hemostatic plugs shown are included in supplemental Video 6. Data are representative of at least 6 injuries in 2 mice.
Discussion
Achieving hemostasis following penetrating injuries is essential for the survival of organisms that possess a closed, high-pressure circulatory system. In the present study, we examined the hemostatic response in detail and found that it produces an organized structure in which different elements of the platelet-signaling network give rise to regions that differ in platelet activation state, packing density, and stability. These differences are illustrated in the model shown in Figure 7. We found that the regional heterogeneity shown in the model emerges shortly after injury and persists for at least 1 hour, which was the length of our observation time. The first signs include the appearance of P-selectin on the platelets nearest the site of injury. At approximately the same time, blood loss ceases and a reduction in the penetration of fluorescent albumin and dextran into the core of the platelet mass becomes apparent.

A model for hemostatic plug formation dictated by the interaction of local conditions with the platelet signaling network. The data presented show that during initial formation and up to 1 hour post-injury, a hemostatic plug is comprised of distinct regions defined by the degree of platelet activation and packing density, as depicted in panel A. The characteristics of each region are indicated in A. We propose that this architecture is a result of a gradient of soluble agonists emanating from the site of injury, as shown in (B). Thrombin generation at the site of injury drives platelet activation in the core region (blue to purple/pink in the figure), but its propagation away from the vessel wall is limited. A gradient of ADP and potentially TxA2 emanating from activated platelets (purple to red in the figure) extends farther and results in the recruitment of additional platelets forming the shell region. Additional platelet signaling, such as contact-dependent signaling pathways, reinforces the architecture via local positive feedback.
A model for hemostatic plug formation dictated by the interaction of local conditions with the platelet signaling network. The data presented show that during initial formation and up to 1 hour post-injury, a hemostatic plug is comprised of distinct regions defined by the degree of platelet activation and packing density, as depicted in panel A. The characteristics of each region are indicated in A. We propose that this architecture is a result of a gradient of soluble agonists emanating from the site of injury, as shown in (B). Thrombin generation at the site of injury drives platelet activation in the core region (blue to purple/pink in the figure), but its propagation away from the vessel wall is limited. A gradient of ADP and potentially TxA2 emanating from activated platelets (purple to red in the figure) extends farther and results in the recruitment of additional platelets forming the shell region. Additional platelet signaling, such as contact-dependent signaling pathways, reinforces the architecture via local positive feedback.
These findings extend those that have appeared previously, mapping regional differences within the hemostatic plug to different parts of the platelet-signaling network and showing that the consequences of stable proximity between activated platelets, which we hypothesized previously, actually do occur.19 The latter include the enabling of contact-dependent signaling and the emergence of barriers to the free penetration of plasma-borne molecules. As noted in the Introduction, we chose to focus on the response to penetrating injuries in the present study, viewing these as being more representative of hemostasis than the thrombotic response seen with injuries based on the use of ferric chloride or rose Bengal.26 Although information about the extent of laser penetration through the vessel wall was not always included, studies from the Furie Laboratory, which pioneered this approach,5,6 and others4,8,27 suggest that the results reported here are not peculiar to the 2 methods used or the tissue studied. For example, the responses we obtained in the cremaster muscle microcirculation (30–45 µm arterioles) are consistent with a prior report of penetrating laser injury in mesenteric arterioles (65–100 µm diameter).8 Based on our studies in mutant mice and with clinically relevant therapeutic agents, we speculate that our findings are also pertinent for pathologic thrombosis.
How might this regionalized structure emerge? A key feature is the extent of platelet activation in the core and shell regions. Platelets in the core are P-selectin positive, meaning that they have undergone α-granule exocytosis; however, this is not their sole defining characteristic. The finding that only P-selectin–negative platelets embolize suggests that firm attachment is dependent on the level of platelet activation, with platelets that have achieved activation states sufficient to cause α-granule secretion being firmly attached to the site of injury. The extent to which platelets in the outer shell are activated is less clear from the present study. Previous reports have noted discoid platelet accumulation following vascular injury in vivo,1-4 suggesting that the outer shell is composed of platelets that are merely tethered. It is unlikely, however, that they are fully quiescent; our studies with the Gi2α(+/G184S) mice and cangrelor indicate that Gi2-dependent signaling regulates the size of the shell.
In addition to α-granule exocytosis, the core region is also marked by a reduction in porosity and an increase in packing density. The relationship of these properties is not necessarily cause and effect. The studies in which fluorescent albumin or dextran were injected prior to or immediately after the injury rather than 20 to 25 minutes later indicate that hemostasis is achieved and porosity reduced before many of the platelets have become P-selectin positive. Increased packing density narrows the gaps between adjacent platelets, presumably reflecting platelet shape change, cohesion, and, eventually, clot retraction. This reduces the penetration of plasma-borne molecules, including those that might otherwise act as thrombin inhibitors or triggers of fibrinolysis, as well as those such as clotting factors that might contribute to additional thrombin generation. Efflux of platelet granule contents and proteolytically shed molecules would likely be similarly affected. If so, then the narrowing gaps between platelets can be viewed as providing shelter for the accumulation of platelet products, including autocrine and paracrine agonists.19 Increased packing density also facilitates contact-dependent signaling, as illustrated by the results obtained with the sema4D knockout mice. Sema4D acts as a contact-dependent amplifier of collagen-induced platelet activation.12,13 As shown here for the first time, deleting it has a much greater effect on the core than on the shell.
Finally, what are some of the factors that result in the observed hierarchical structure and how do they relate to the platelet-signaling network, which is presumably common to all platelets in the hemostatic plug? We propose that regional differences in platelet activation reflect regional differences in the distribution of platelet agonists and that, as a result, different elements of the platelet-signaling network are invoked (Figure 7). Although collagen is limited to the vascular wall, thrombin, ADP, and thromboxane A2 (TxA2) are not. The observed distribution of fibrin implies that thrombin generation occurs primarily in the region of the core nearest the site of injury, as do our recent studies with a thrombin sensor tethered to the platelet surface.18 As thrombin diffuses outward, it would be expected to encounter potent inhibitors, limiting its impact on platelets farther from the vessel wall. This localization of thrombin results in robust platelet activation that is spatially confined, thus forming the core region. Consistent with this model, hirudin prevented core formation but left a residue of the platelet shell.
TxA2 and ADP, on the other hand, are small enough to diffuse outward from the region where platelets are sufficiently activated to produce them and their effects are limited by short half-life (TxA2), hydrolysis (ADP), and dilution. The data obtained with the Gi2α(+/G184S) mice and cangrelor suggest that ADP promotes the growth of the shell. Notably, the Gi2α gain of function mutation and cangrelor produce opposite effects, the former increasing and the latter decreasing the size of the shell. Neither intervention appears to affect the initial rate of platelet accumulation (compare Figure 5C and Figure 6A). By implication, a decline in the local ADP concentration below a critical level may be a major factor in limiting the size of the shell and preventing unwarranted vascular occlusion following limited injury. This may help to account for the ability of P2Y12 antagonists, such as cangrelor and clopidogrel, to reduce platelet accumulation without always causing bleeding. Aspirin is similarly effective at reducing unwarranted platelet accumulation without major hemostatic deficiency, suggesting TxA2 may also regulate the shell region, although this was not examined here. Note that the gradients of thrombin, ADP, and TxA2 that we illustrate in Figure 7 need not arise from precisely the same location nor extend the same distances from the vessel wall. Thrombin appears to be generated close to the site of injury and to not extend the full width of the core region before it declines below critical concentration. ADP release from platelet-dense granules, on the other hand, presumably occurs throughout the core and potentially in platelets that have not undergone α-granule secretion as well. This supports the idea that the ADP gradient extends well into the shell.
In conclusion, the results presented here provide a detailed analysis of the hemostatic response to penetrating injury made possible by improvements in intravital imaging technologies and relate the findings to regional differences in agonists, signaling, and packing density. The organization that was observed demonstrates that hemostatic plugs are heterogeneous in composition and suggests that a hierarchical structure serves both to stabilize the hemostatic plug and limit its growth. The results also show how local conditions and different elements of the platelet-signaling network can work together to produce regional differences in the platelet activation state and regulate the hemostatic response to injury. Thus, what has long seemed like a surfeit of signaling pathways in platelets can be rationalized by the differential contribution of these pathways, which depend upon location of the platelets within the hemostatic mass in vivo. Although antiplatelet agents were used primarily as tools to understand the hemostatic response, the results provide a new context for understanding their clinical impact and offer new insights into how future agents might be designed.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the American Heart Association (11SDG5720011 to T.J.S.) and the National Institutes of Health: National Heart, Lung and Blood Institute (HL40387 to L.F.B., and HL103419 to S.L.D. and L.F.B). The confocal intravital microscopy system employed was partially funded by NIH-NCRR shared instrument grant 1S10RR26716-1.
National Institutes of Health
Authorship
Contribution: T.J.S. designed and conducted experiments, analyzed data, and wrote the manuscript. J.W., E.A.T., and S.L.C. conducted experiments and analyzed data. K.M.W., R.V., and S.L.D. analyzed and interpreted data. L.F.B. designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Timothy J. Stalker, PhD, University of Pennsylvania, Perelman School of Medicine, 809 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: tstalker@mail.med.upenn.edu, and Lawrence F. Brass, MD/PhD, University of Pennsylvania, Perelman School of Medicine, 809 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: brass@mail.med.upenn.edu.

