

Abstract
Ephrin receptors (Eph) have been implicated to contribute to cancer pathogenesis in a way by which receptors can both promote or suppress cancer progression depending on their cellular contexts. Mechanism for Eph receptor tumor suppressor functions are unknown. In a previous study, we found that pediatric acute myeloid leukemia (AML) sample lysates showed a significant decreased EphB1 peptide activity in comparison to normal bone marrow (NBM). In this study, we challenged to investigate EphB1 receptor transcriptional regulation and biological insights in AML.
Quantitative RT-PCR analysis and bisulfate pyrosequencing was used to examine mRNA expression and methylation in AML cell lines and AML patient samples. AML cell proliferation (cell counts) and apoptosis (annexin V/PI) were analyzed after EfnB1 ligand stimulation. We generated constitutive EphB1 overexpression constructs in AML cell lines. Downstream in vitro effects were studied using quantitative RT-PCR, flowcytometry, phospho-proteome arrays, chromatin immunoprecipitation (ChIP), and immunoblot analysis.
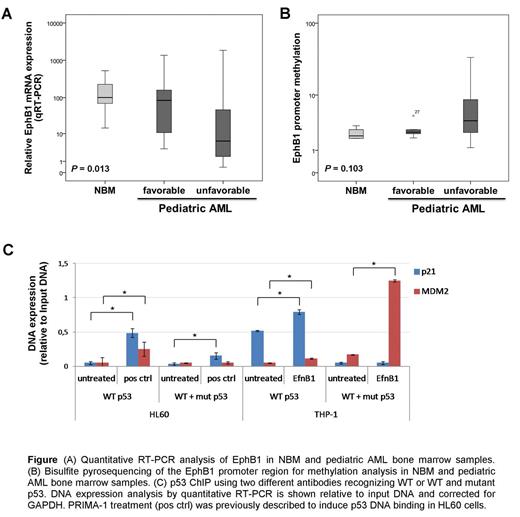
First, we examined EphB1 mRNA expression and methylation in primary pediatric AML samples, which revealed that EphB1 mRNA expression was significantly lower in unfavorable AML cytogenetic subgroups as compared to favorable AML patient samples (Fig. A, P = 0.013). The EphB1 promoter region was found increasingly methylated in approximately 20% of the pediatric AML patient samples (n=21), but not in NBM (n=5, Fig. B). In AML cell lines, we confirmed that EphB1 promoter methylation resulted in epigenetic gene silencing (P = 0.010).
As a next step we challenged to study the biological relevance of EphB1 suppression. EfnB1 stimulation of EphB1high THP-1 cells inhibited the cell proliferation and induced apoptosis, while EphB1low HL60 and MOLM13 cells remained unaffected upon EfnB1 stimulation. Cell cycle analysis revealed that EfnB1 stimulation mediated phenotypic effects in EphB1high THP-1 cells could be assigned to a G2/M cell cycle arrest. The cell cycle arrest and the induction of apoptosis as a common consequence was confirmed by phospho-proteome, immunoblot, and qRT-PCR analysis. We found an induction of DNA damage control protein activation and cell cycle inhibitors Chk-2, p21, p27, p53, and CDC2tyr15, and a reduction of cell survival proteins HSP27 and BCL-2 accompanied by an increase of Bax. Next, we investigated whether EfnB1 stimulation of THP-1 could have restored the DNA damage control system by increasing DNA binding of mutant and wildtype p53. Performing ChIP assays using p53 antibodies recognizing wildtype or both mutant and wildtype p53 followed by p21 and MDM2 quantitative RT-PCR analysis revealed that EfnB1 stimulation of THP-1 cells significantly increased p53 DNA binding (Fig. C).
Introducing a constitutive EphB1 overexpression into EphB1 methylated HL60 and MOLM13 cell lines resulted in an instant increase in apoptosis relatively to empty vector control cells. As expected, G2/M cell cycle inhibiting phosphorylation of CDC2tyr15 was upregulated. EfnB1 stimulation of the GFP sorted remaining viable cells showed to decrease the AML cell survival in EphB1 overexpressed AML cells as compared to empty vector controls. Additionally, we found that EphB1 demethylation by 5-Aza-2'-deoxycytidine was able to restore EphB1 membrane protein expression in EphB1 methylated AML cells and simultaneously reduced the AML cell survival.
In this study, we found a common downregulation of EphB1 mRNA that was significantly associated with an unfavorable prognosis in pediatric AML. The downregulation of EphB1 could be explained by EphB1 promoter hypermethylation in a subset of AML samples. Downregulation of EphB1 was found to be one of the mechanisms through which AML cells suppress p53 DNA binding that impairs the DNA damage control system to support a proliferative and anti-apoptotic AML cell survival advantage.
No relevant conflicts of interest to declare.

