

Key Points
No difference in survival, early toxicity, or occurrence of tumors between ARTEMIS and RAG-deficient SCID.
ARTEMIS-deficient patients develop late toxicity following HCT: growth retardation, endocrinologic deficiencies, and dental abnormalities.
Abstract
A subgroup of severe combined immunodeficiencies (SCID) is characterized by lack of T and B cells and is caused by defects in genes required for T- and B-cell receptor gene rearrangement. Several of these genes are also involved in nonhomologous end joining of DNA double-strand break repair, the largest subgroup consisting of patients with T−B−NK+SCID due to DCLRE1C/ARTEMIS defects. We postulated that in patients with ARTEMIS deficiency, early and late complications following hematopoietic cell transplantation might be more prominent compared with patients with T−B−NK+SCID caused by recombination activating gene 1/2 (RAG1/2) deficiencies. We analyzed 69 patients with ARTEMIS and 76 patients with RAG1/2 deficiencies who received transplants from either HLA-identical donors without conditioning or from HLA-nonidentical donors without or with conditioning. There was no difference in survival or in the incidence or severity of acute graft-versus-host disease regardless of exposure to alkylating agents. Secondary malignancies were not observed. Immune reconstitution was comparable in both groups, however, ARTEMIS-deficient patients had a significantly higher occurrence of infections in long-term follow-up. There is a highly significant association between poor growth in ARTEMIS deficiency and use of alkylating agents. Furthermore, abnormalities in dental development and endocrine late effects were associated with alkylation therapy in ARTEMIS deficiency.
Introduction
A subgroup of patients with severe combined immunodeficiencies (SCID) lacks both T and B cells while natural killer (NK) cells are normal. In these T−B−NK+SCID patients, a number of defects have been found in genes which are essential for V(D)J recombination. This recombination process allows the generation of functional and highly diverse immunoglobulin and T-cell receptor genes. If deficient, the maturation of B and T lymphocytes is arrested. Mutations have been found in the recombination activating complex composed of the recombination activating gene 1 (RAG1), RAG2, and high-mobility group protein 1 (HMG1) proteins, and in some of the factors of the nonhomologous end joining (NHEJ) pathway of DNA double-strand break repair, for example, the nuclease DCLRE1C/ARTEMIS, XLF/CERNUNNOS, the ligase complex XRCC4/DNA LIGASE IV and PRKDC/DNA-PKcs.1 The majority of V(D)J recombination defects are caused by mutations in RAG 1 or 22,3 and in DCLRE1C/ARTEMIS.4,5
Whereas RAG1 and RAG2 are highly regulated and protein is found in lymphoid progenitors only, factors of the NHEJ pathway are ubiquitously expressed. Because defects of NHEJ result in genomic instability due to impaired DNA repair as reflected by increased in vitro cellular radiosensitivity, they are associated with an increased potential for oncogenic translocations6 and risk of malignant transformation.7 In addition, tissue damage as induced by radiochemotherapy in the context of hematopoietic cell transplantation (HCT) may be poorly tolerated, as noted in other disorders due to DNA repair defects, such as in Fanconi anemia and Nijmegen breakage syndrome.8-12
In fact, a report of 18 ARTEMIS-deficient SCID patients of Native American descent found poorer outcome and lack of secondary teeth in those patients treated with alkylating agents.13 Also, analysis of 90 SCID patients with different molecular defects found that patients with ARTEMIS deficiency were at higher risk for late complications.14 We therefore compared patients with RAG1/2 and ARTEMIS-deficient SCID with respect to outcome post-HCT focusing on survival, acute complications (toxicity, acute graft-versus-host disease [GVHD]), and late complications (chronic GVHD, growth and development, secondary malignancies, endocrinologic sequelae). We also analyzed long-term immunologic outcome.
Material and methods
Patients
Data from the medical records of all patients with genetically confirmed ARTEMIS and RAG deficiencies who received transplants between 1985 and 2009 at the University of California, San Francisco Benioff Children’s Hospital, Hôpital Necker–Enfants Malades, France (Paris), and at the Department of Pediatric and Adolescent Medicine, University Medical Center Ulm, Germany (Ulm) were collected by each center, combined, and analyzed retrospectively in a multicenter study. Informed consent from the parents for data collection and analysis had been obtained upon hospital admission in accordance with the Declaration of Helsinki. Retrospective data collection was in accordance with the ethical committees of the 3 centers. Data collection included clinical data upon presentation, genetic mutations, HCT characteristics, engraftment, chimerism studies, immunologic reconstitution, and late clinical outcome. Clinical follow-up post-HCT focused on acute organ toxicity, acute and chronic GVHD, secondary malignancies, and persistent medical problems following HCT.
Early complications were defined as all clinical events occurring during the first 2 years post-HCT including early death, toxicity, acute GVHD, and systemic infections.
Late complications were defined as all clinical events occurring later than 2 years post-HCT, that is, late death, late infections, chronic GVHD (cGVHD), autoimmune or autoinflammatory complications, nonautoimmune endocrinologic disorders, dental abnormalities, poor growth as well as occurrence of malignancies.
The following definitions were used for preconditioning regimens and additional transplant procedures. Myeloablative preconditioning regimen was defined as treatment with busulfan at either 16 mg/kg orally or 12.5 mg/kg intravenously associated with either cyclophosphamide or fludarabine. The use of a lower dose of busulfan at 8 mg/kg orally was defined as nonmyeloablative conditioning. A minority of patients (n = 13/145, 9%) received immunosuppressive conditioning using fludarabine or cyclophosphamide without busulfan. The term of “alkylating agent” is used for any type and dosage of an alkylating chemotherapeutic substance, that is, any of the following: busulfan, treosulfan, thiothepa, melphalan, or cyclophosphamide. For patients having received an additional procedure, a retransplant procedure required prior myeloablation, otherwise it was considered a stem cell boost.
Immunologic data were obtained at each visit in outpatient clinics and included T-cell phenotyping, T-cell function, chimerism of whole-blood and leukocyte subpopulations, including granulocytes, mononuclear cells (MNCs), and CD3+ vs CD3− T-cell fractions, using the following markers: chromosomal constellation XX/XY with fluorescent in situ hybridization, short tandem repeat analysis, or HLA antigens detected in fluorescence-activated cell sorter analysis.
Myeloid engraftment was defined as donor myeloid chimerism >10%, measured either in granulocytes or in CD34+ bone marrow precursors. Normal immune reconstitution at ≥2 years post-HCT was defined as CD4+ T cells >600/µL.15 As data for vaccination antibody titers was not available for all patients, persisting need for immunoglobulin replacement therapy was used as a surrogate indicator for poor B-cell reconstitution.
Survival analysis and early complications (<2 years post-HCT) were performed on the whole cohort of 145 patients. Late outcome and quality of immune reconstitution were studied in patients alive 2 years post-HCT and with a follow-up >2 years (n = 92). For growth analysis, only patients with a follow-up >5 years following HCT were included. SD scores and z-scores for height were calculated from Centers for Disease Control and Prevention (CDC) growth charts at 2 to 2.5 years post-HCT and at the latest available time point. Height SD scores were calculated using least mean square transformation as described by Cole.16
Statistical analysis
Population characteristics of the overall cohort and of the late cohort were presented and compared at baseline using the Fisher exact test and a nonparametric Kruskal-Wallis test. Survival analysis was then performed using Cox modeling to identify risk factors of death. The end point was the time of death. Factors included in the model were (1) pre-engraftment data such as molecular diagnosis, Omenn syndrome, viral infection, ulcers, (2) characteristics of type of conditioning regimen and HCT, and (3) early features of HCT data (early complications, myeloid chimerism, additional post-HCT procedures). All variables were entered into the multivariate Cox model, and we used a backward stepwise selection technique, with a significance limit of P > .05 set for removal from the model. Proportionality assumption of the Cox proportional hazard model was tested using Schoenfeld and scaled Schoenfeld residuals tests. Early and late immune reconstitution (CD4+ T-cell count at 1, 2, and 5 years post-HCT, and requirement for intravenous immunoglobulins (IVIG) at last follow-up) were studied by logistic regression models, including a continuous time variable. Risk factors associated with late complications occurrence (all complications mixed, and then analyzed separately: autoimmunity and infection, damage, and teething problems) in the cohort with >2 years of follow-up were studied using Poisson modeling. The same variables were used as in Cox analysis, and the final model was built by a stepwise procedure. For all modeling (Cox, logistic regression, and Poisson) patients were clustered using a group variable in the model to take into account between-center variability. Statistical analyses used Stata/IC 12.0 for Mac software (StataCorp).
Results
Patients’ characteristics and details of HCT
A total of 145 patients (ARTEMIS deficiency, n = 69; RAG1/2 deficiencies, n = 76) were entered into the study. Athabascan-speaking Native Americans, a population carrying a founder mutation in exon 8 of DCLRE1C leading to a stop codon and to a lack of ARTEMIS protein expression, accounted for 28% (19 of 69) of ARTEMIS-deficient patients.17 A large percentage (34%, 26 of 76) of RAG 1- or 2-deficient patients were of North African origin. RAG1 mutations were present in 63% (48 of 76), and RAG2 mutations in 37% (28 of 76) (for details, see supplemental Table 1, available on the Blood Web site). Omenn phenotype was almost exclusively seen in RAG1/2-deficient patients (P = .001).18 Characteristics of patients at presentation as well as details of treatments such as donor choice, use of preconditioning regimen and type of conditioning, were not significantly different between patients with RAG1/2 and with ARTEMIS mutations (Table 1).
As compared with the other ARTEMIS-deficient patients, Athabascan-speaking Native Americans patients suffered more frequently from orogenital ulcers (P < .05)19 and less frequently from viral infections prior to their first HCT (P < .01). They also received busulfan-containing conditioning regimens less often (n = 4/23, 17% vs n = 19/46, 41%, P < .05). Overall exposure to alkylating agents, including immunosuppression with cyclophosphamide only, however, was comparable in both groups of ARTEMIS-deficient patients (data not shown).
Outcome according to donor and conditioning
Donors in 42 of the 145 were HLA-identical family members (ARTEMIS, n = 17; RAG, n = 25), either siblings (in 24 cases) or close relatives, usually parents. These patients received unmanipulated grafts without myeloablative conditioning with the exception of 3 cases with SCID complicated by Omenn syndrome. HLA-identical family donor transplantation resulted in T-cell reconstitution albeit with CD4+ T-cell numbers in a lower range in the majority of patients. Thirty-seven of the 42 patients (88%) survive long-term without recurrent or severe infections. Outcome was comparable in ARTEMIS and RAG-deficient patients (Figure 1, Tables 2 and 3).

Kaplan-Meier estimates of survival for SCID patients with RAG or ARTEMIS deficiencies according to donor.
Kaplan-Meier estimates of survival for SCID patients with RAG or ARTEMIS deficiencies according to donor.
Reconstitution of B-cell immunity, which in B−SCID always reflects donor B-cell development, was observed less consistently: 21 of 37 patients developed B cells without preconditioning, the others remain on regular immunoglobulin substitution. Repeat transplants were required in 3 cases to overcome aplastic anemia developing after initial transplantation, and resulted in myeloid donor cell chimerism, which was only exceptionally observed in other nonconditioned cases (see Table 2).
The majority of patients (n = 82) underwent T-cell–depleted HLA-haploidentical family donor transplantation (ARTEMIS, n = 43; RAG, n = 39), using T-cell depletion for GVHD prevention. Of these 82 cases, 17 cases were transplanted without conditioning chemotherapy and 15 cases with cyclophosphamide or fludarabine with variably added serotherapy (ATG, anti-LFA1/anti-CD2). The outcome of HCT in these 32 cases was generally poor, with complete graft failure or insufficient T-cell reconstitution representing a predominant problem regardless of the underlying molecular diagnosis (see Table 2). Only 8 patients engrafted (4 after no pretreatment, 4 after cyclophosphamide) and developed sustained T-cell immunity, but none developed B cells. Most of the patients with graft failure (18 of 22) underwent repeat HCT using the same donors except 2 cases transplanted from matched unrelated donor (MUD), in the majority following conditioning with busulfan. Repeat HCT was successful in 9 of 18 cases including 2 cases retransplanted from MUD (data not shown).
In the remaining 46 of 82 cases undergoing HLA-haploidentical transplantation, conditioning was with busulfan which was used in combination either with cyclophosphamide or with fludarabine (see Table 2). Thirty-one of these patients survive with T-cell reconstitution (9 of 14 after low busulfan, 22 of 32 after high busulfan), and most also developed B cells (6 of 9 in the former and 18 of 22 in the latter group). The survival rate in all patients undergoing HCT with busulfan, either for initial or for repeat HLA-haploidentical HCT, was 63% (36 of 55), and was similar in RAG and ARTEMIS patients (Figure 1). Donor myeloid cell chimerism was commonly observed, regardless of the dosage of busulfan used. In several patients with absent B-cell engraftment, boost transplants were explored, but failed to be effective. A summary of all HLA-haploidentical transplants is shown in Table 2, including 4 further patients in whom busulfan was replaced by treosulfan (n = 3) or thiotepa (n = 1) for conditioning. Of the remaining 21 patients analyzed in this study, 8 patients (of whom 5 are long-term survivors) received MUD transplants following myeloablative conditioning (with 1 exception). Thirteen patients were transplanted without conditioning from HLA-mismatched (1 antigen) family donors and without T-cell depletion. Six of these patients survive, while 7 died of complications related to GVHD (Table 2).
Comparative analysis of survival and early complications in ARTEMIS- and RAG-deficient patients
Overall survival was 62% (95% CI, 52-70). Molecular diagnosis did not significantly influence survival in univariate and multivariate analysis whereas donor type did with HLA identical donors having a better outcome than haploidentical donors who received myeloablative conditioning (Figure 1, Table 2). Factors associated with mortality in multivariate analysis were presence of viral infection prior to HCT, age at diagnosis >3 months, presence of cGVHD, and need for retransplantation. The use of myeloablative conditioning regimen did not influence survival. Outcome of patients with Omenn phenotype (n = 14 with RAG deficiencies, n = 1 with DCLRE1C deficiency) in univariate and multivariate analyses was comparable to the rest of the cohort (data not shown). Deaths occurred mostly during the first 2 years post-HCT (median time to death was 5 months following HCT). Causes of death were not different in ARTEMIS and RAG1/2-deficient SCID patients (supplemental Table 2), with infections being the most frequent cause. There was no difference in the incidence of severe mucositis, veno-occlusive disease/sino-obstructive syndrome or acute GVHD (data not shown).
Comparative analysis of long-term immune reconstitution and late complications in ARTEMIS- and RAG-deficient patients
Immune reconstitution and long-term clinical outcome was analyzed in 92 patients who survived at least 2 years after HCT, excluding 8 surviving patients because of incomplete follow-up data. Characteristics of this late cohort and HCT characteristics are shown in supplemental Table 3. Median follow-up was 8.5 years (range, 2-28 years). Surviving patients with RAG1/2 SCID had received myeloablative conditioning regimens more frequently compared with ARTEMIS-deficient SCID (P = .04). Distribution of donor origin was also significantly different with more transplants from haploidentical donors in ARTEMIS-deficient SCID patients.
CD4+ T-cell numbers at 2 years after HCT were normal (>600/µL) in 61% of patients. The main independent predictive factors of a normal CD4+ T-cell count in univariate and multivariate analysis were the use of myeloablative conditioning and the presence of donor myeloid chimerism (Table 4). Although there was no difference between RAG1/2 and ARTEMIS-deficient patients using univariate or multivariate analysis in the CD4+ count, being of Native American descent was associated with a lower CD4+ T-cell count in univariate analysis.
Forty-two of 89 patients (47%) required immunoglobulin substitution at last follow-up. In univariate and multivariate analysis, SCID patients with ARTEMIS deficiency, but also poor T-cell reconstitution, requirement of an additional transplant procedure and HCT from a haploidentical donor predisposed for long-term requirement of immunoglobulins (Table 5).
A total of 49% (45 of 92) of the patients had 115 clinical events during follow-up as shown in Table 6 for ARTEMIS and RAG-deficient patients, respectively. None of the patients has so far presented with malignant disease. A total of 22 patients developed 26 autoimmune manifestations, commonly associated with persistent cGVHD. Twenty-two patients (24%) developed 24 severe or recurrent infections during follow-up. Poor growth and requirement for nutritional support were reported in 27 (29%) and 12 (13%) patients, respectively. Additional noninfectious and nonautoimmune complications occurred exclusively in 7 patients with ARTEMIS deficiency (13 events) as follows: central growth hormone deficiency (n = 4), central hypothyroidism (n = 3), insulin-dependent diabetes (n = 2), renal tubulopathy (n = 2), pancreatic exocrine insufficiency (n = 1), and pulmonary fibrosis (n = 1). Median age of patients with these complications was 15 years (range from 10 to 20 years). Abnormal development of permanent teeth was also exclusively observed in patients with ARTEMIS deficiency (n = 10). In univariate analysis, a conditioning regimen with alkylating agents was strongly associated with these late complications (P = .002, Table 7). Indeed, among ARTEMIS patients with a follow-up >5 years, noninfectious/nonautoimmune problems and dental abnormalities were found exclusively in individuals who received alkylators, respectively, in 30% and 48% of this subgroup of patients.
An in-depth univariate and multivariate analysis of late complications, where all clinical events were considered (cGVHD, autoimmunity, infections, noninfectious and nonautoimmune miscellaneous complications, dental abnormalities, growth retardation, and requirement for nutritional support) revealed the following predictors: molecular diagnosis of ARTEMIS-deficient SCID (P < .0001), treatment with alkylating agents (P < .0001), and quality of immune reconstitution (defined by the necessity of a boost/retransplantation and IVIG replacement) (Table 8).
Nonmyeloablative conditioning and poor immune reconstitution (requirement for an additional procedure and for IVIG) were associated with late immunodysregulatory problems and late infections (supplemental Table 4a).
Factors predictive of poor growth and requirement for nutritional support >2 years post-HCT in multivariate analysis were ARTEMIS deficiency, presence of viral infection prior to HCT, myeloablative conditioning regimen, and use of alkylating agents (supplemental Table 4b). Among patients aged >5 years who received alkylators, 86% (18 of 21) of ARTEMIS and 12% (2 of 17) of RAG-deficient patients had growth deficiency. Available growth charts of patients aged >5 years at the time of this analysis were compared between ARTEMIS- (n = 30, aged 6-20 years; median, 13 years) and RAG- (n = 20, aged 6-26.5 years; median, 14 years) deficient patients. Patients with ARTEMIS-deficient SCID having received a conditioning regimen with alkylating agents showed a significant decrease in height at the last follow-up visit compared with patients who had not received alkylating agents (P < .03 for girls, P < .0001 for boys). Boys with ARTEMIS deficiency following therapy with alkylating agents had the greatest loss in height in comparison with male patients without this therapy (Figure 2).
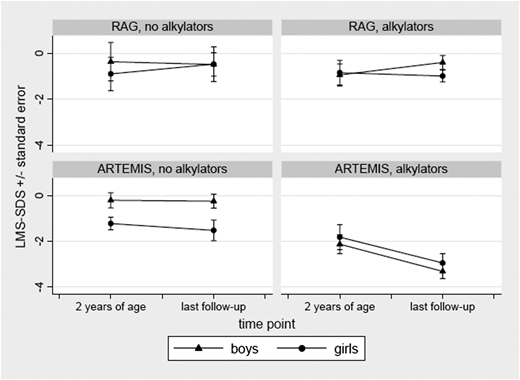
Comparison of height standard deviation scores for ARTEMIS vs RAG-deficient SCID patients at 2 years of age and at last follow-up. (Left panels) No chemotherapy with alkylators. (Right panels) Following therapy with alkylators.
Comparison of height standard deviation scores for ARTEMIS vs RAG-deficient SCID patients at 2 years of age and at last follow-up. (Left panels) No chemotherapy with alkylators. (Right panels) Following therapy with alkylators.
Discussion
A curative treatment approach in patients with SCID aims at full long-term immune reconstitution of T- and B-cell functions and frequently includes the use of conditioning regimens.15 A transplant regimen omitting conditioning is more likely to result in persistent B-cell deficiency and occasionally poor T-cell reconstitution, in particular, in HLA-haploidentical transplants. The decision whether or not to use a chemotherapeutic conditioning regimen for optimal T- and B-cell engraftment is more difficult in radiosensitive forms of SCID. Prior to this study it was not clear whether HCT in patients with ARTEMIS deficiency, when performed with conditioning, is associated with higher tissue toxicity, more severe GVHD, and secondary tumors as has been observed in patients with other radiation-sensitivity syndromes such as Nijmegen breakage or Fanconi anemia following HCT.10,11 A single-center study including all transplanted SCID patients with a median follow-up of 14 years following HCT14 found a striking difference in event-free survival between SCID patients with ARTEMIS deficiency and SCID due to other genetic defects including RAG1/2 deficiencies. Also, a poor clinical outcome in long-term survivors as well as reduced final height and dental abnormalities has been reported in Athabascan-speaking Native Americans with ARTEMIS-deficient SCID, all with the same homozygous mutation.13 These findings prompted us to reinvestigate and compare the outcomes of HCT in patients with ARTEMIS- and RAG1/2-deficient SCID in a large multicenter cohort.
ARTEMIS- and RAG-deficient SCID do not differ in survival, early toxicity, or occurrence of tumors following HCT but develop more late complications after HCT
In our study, a comparative analysis of early outcome between patients with ARTEMIS- and RAG-deficient SCID revealed that these 2 groups do not differ in survival (Figure 1), nor in incidence of acute organ toxicity (mucositis, veno-occlusive disease) and GVHD. As a key factor in the NHEJ pathway, the ARTEMIS protein is ubiquitously expressed and has a central role in DNA double-strand break repair. Therefore, our observations are surprising in that more acute toxic damage from preconditioning chemotherapy and radiation could have been expected in ARTEMIS-deficient SCID compared with patients with RAG deficiencies. However, in long-term follow-up, ARTEMIS patients showed significantly more late complications compared with RAG patients. Use of alkylating agents and poor immune reconstitution was also related to poor outcome in the patients with ARTEMIS-deficient SCID. These complications consisted of infections, immunodysregulation, noninfectious/nonautoimmune endocrinologic dysfunction, growth problems, and permanent teeth abnormalities. No malignancy occurred in any patient during a mean follow-up of 10 years (range, 2-28 years post-HCT). Longer follow-up of patients with ARTEMIS-deficient SCID following HCT is needed to confirm that the risk of tumor development is indeed not elevated compared with nonradiosensitive SCID variants.
ARTEMIS-deficient patients conditioned with alkylating agents develop specific problems not observed in RAG deficiency and have a prepubertal decrease in growth velocity
ARTEMIS-deficient patients developed specific late problems after HCT: dental abnormalities, endocrinologic deficiencies, renal tubulopathy, and pancreas exocrine deficiency were exclusively encountered in this group of patients; growth retardation was significantly more frequent in ARTEMIS patients as compared with RAG1/2-deficient patients. The main factor associated with these complications was exposure to alkylating. These striking observations deserve further comments.
The effect of radiotherapy on growth and endocrine function is well documented20-23 for pediatric cohorts with malignant diseases whereas the effects of a busulfan/cyclophosphamide-based conditioning regimen on growth and endocrine function in pediatric recipients are controversial.22 A multicenter study examining final height in 181 pediatric patients following HCT for various hematologic disorders concluded that conditioning with busulfan and cyclophosphamide had no significant effect on final height achievement.20 The authors identified 2 major risk factors for decreased final height: male gender and young age at HCT following combined treatments including chemotherapy and irradiation. In our study, when compared with normals, we found that patients with SCID due to RAG1/2 deficiencies showed no difference in height at last follow-up whether treated with or without alkylating therapy. In contrast, in patients with ARTEMIS-deficient SCID following therapy with alkylators, we found a significant long-term height loss, especially in boys. Interestingly, patients with ARTEMIS deficiency who did not receive alkylators had normal growth, comparable to those with RAG defects. The mechanisms of growth failure associated with exposure to alkylating agents in ARTEMIS-deficient patients remain undefined and require further study.
Dental problems have previously been described in patients of Native American background with ARTEMIS-deficient SCID13 and in 3 transplanted SCID patients of unknown molecular diagnosis.24 Dental irregularities (mainly microdontia, hypodontia, abnormal roots, enamel hypoplasia, and missing secondary teeth) are reported in pediatric recipients of alkylating therapy for neoplasias and in patients treated with radiotherapy.25,26 Young age at the time of treatment appeared as an additional risk factor. In the present analysis, these abnormalities were only documented in SCID patients due to ARTEMIS, but not RAG1/2 deficiencies.
The effects of alkylating agents on growth and teething problems do not seem to be related to enhanced mutagenesis. The combination of ARTEMIS deficiency with low repair potential and damage through alkylating agents may lead to increased apoptosis in susceptible tissues such as endocrine glands and developing tooth buds necessary for normal calcification and coronal and root formation. Induced changes in methylation patterns or impairment of transcription of damaged DNA may lead to altered gene expression with potential functional consequences in susceptible tissues (eg, ameloblasts, cementoblasts, odontoblasts, hypophysis, islet cells, testicles, or ovaries).
Importantly, late complications following conditioning with alkylating agents in patients with ARTEMIS-deficient SCID often occur many years post-HCT. Therefore, these patients warrant thorough clinical follow-up including regular endocrinologic evaluation. Patients presenting with hormone deficiencies following HCT might benefit from hormone substitution including thyroid and growth hormone treatment.
Dental abnormalities may lead to functional problems requiring dental appliances and prosthetic correction. Some of these developmental defects may be underrecognized by both patients and physicians. Therefore, close follow-up should also include a thorough dental evaluation at regular intervals.
Myeloablation is needed for full immunologic reconstitution
Infections and immunodysregulatory complications are mainly related to the quality of immune reconstitution. In multivariate analysis, the use of myeloablation correlated with higher CD4+ T-cell counts and less need for immunoglobulin replacement therapy, which is consistent with previous reports.14,15,27-29 Native American patients, who received less myeloablation compared with other ARTEMIS and RAG1/2 patients had a trend for poorer immune reconstitution (lower CD4+ T cells and a higher rate of immunoglobulin requirement in univariate analysis) consistent with the high rate of late infections (primarily sinusitis) observed in this group of patients.
Conclusion
Myeloablative conditioning clearly supports complete immunologic reconstitution, that is, T- and B-cell engraftment. However, use of alkylating agents in patients with ARTEMIS-deficient SCID carries the risk of significant long-term toxicity including growth retardation, late-onset endocrinologic deficiencies, and dental abnormalities. Similar long-term toxicities were not observed in patients with RAG-deficient SCID. Physicians treating patients with ARTEMIS-deficient SCID need to explain the risks and benefits of conditioning with alkylating agents to the parents so that an informed decision can be made. Regarding long-term immune functions, patients with ARTEMIS deficiency receiving myeloablative conditioning before HCT have a favorable outcome but at the cost of an increase in noninfectious complications. There is a major need for alternative safe conditioning regimens to achieve multilineage engraftment without the long-term toxicity observed in patients with ARTEMIS-deficient SCID prior to conventional HCT, as well as treatment with genetically modified autologous stem cells.
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Dr Diana Hu from the Tuba City Regional Health Care Corporation for her thoughtful and important comments and suggestions. We thank Sandra Steinmann for excellent data management and Prof G. Speit for helpful discussions. We thank the patients’ families, and the staff of the pediatric transplant wards, for their dedication in patient care.
This work was supported by a grant from the Jeffrey-Modell-Foundation. German Federal Ministry for Education and Research (BMBF) grants to K.S. and M.H. (BMBF 01GM0895; BMBF 01GM1111F) and National Institutes of Health, National Institute of Allergy and Infectious Diseases to C.C.D. and M.J.C. (U54 AI82973 and R13 AI116784) are greatly appreciated.
Authorship
Contribution: C.S., B.N., and C.C.D. designed the research, collected data, and wrote the manuscript; C.S. and B.N. analyzed data; M.J.E. and S.L. conducted statistical analyses; K.S., U.P., C.P., J.-P.d.V., and D.M. performed genetic analyses; U.P. reclassified genetic data; C.S., B.N., C.C.D., A.S.S., M.H., M.S.-S., S.A.G., S.B., C.P., B.N.H., D.M., and K.-M.D. contributed to patients’ clinical care; C.P. performed immunologic analyses pre- and post-HCT; M.C. directed the quality of stem cell transplantation; C.D. conceptualized analysis and presentation for patients' growth data; K.-M.D. designed research; and W.F., A.F., and M.J.C. contributed to study design, writing of the report, and patients’ clinical care.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Catharina Schuetz, Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm; e-mail: catharina.schuetz@uniklinik-ulm.de; Benedicte Neven, Unité d’Immuno-Hématologie, Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, France; e-mail: benedicte.neven@nck.aphp.fr; and Christopher C. Dvorak, UCSF Benioff Children’s Hospital, 505 Parnassus Ave, San Francisco, CA 94143; e-mail: dvorakc@peds.ucsf.edu.

