

Key Points
White individuals have a high frequency of the common PAR4 gene (F2RL3) variant Ala120; blacks have a high frequency of Thr120.
PAR4 Thr120 induces greater signaling and is associated with greater platelet aggregation and reduced inhibition by a PAR4 antagonist.
Abstract
Human platelets express 2 thrombin receptors: protease-activated receptor (PAR)-1 and PAR4. Recently, we reported 3.7-fold increased PAR4-mediated aggregation kinetics in platelets from black subjects compared with white subjects. We now show that platelets from blacks (n = 70) express 14% more PAR4 protein than those from whites (n = 84), but this difference is not associated with platelet PAR4 function. Quantitative trait locus analysis identified 3 common single nucleotide polymorphisms in the PAR4 gene (F2RL3) associated with PAR4-induced platelet aggregation. Among these single nucleotide polymorphisms, rs773902 determines whether residue 120 in transmembrane domain 2 is an alanine (Ala) or threonine (Thr). Compared with the Ala120 variant, Thr120 was more common in black subjects than in white subjects (63% vs 19%), was associated with higher PAR4-induced human platelet aggregation and Ca2+ flux, and generated greater inositol 1,4,5-triphosphate in transfected cells. A second, less frequent F2RL3 variant, Phe296Val, was only observed in blacks and abolished the enhanced PAR4-induced platelet aggregation and 1,4,5-triphosphate generation associated with PAR4-Thr120. PAR4 genotype did not affect vorapaxar inhibition of platelet PAR1 function, but a strong pharmacogenetic effect was observed with the PAR4-specific antagonist YD-3 [1-benzyl-3(ethoxycarbonylphenyl)-indazole]. These findings may have an important pharmacogenetic effect on the development of new PAR antagonists.
Introduction
Race/ethnicity is an important factor in determining the outcome of coronary heart disease (CHD). Compared with patients of white race/ethnicity, blacks have a 2-fold higher incidence of CHD, and black race/ethnicity is an independent predictor of worse survival after CHD events, even when confounding demographic, socioeconomic, and clinical factors are considered.1,2 Acute coronary events such as myocardial infarction occur when an occlusive platelet thrombus forms at the site of a ruptured atherosclerotic plaque. There is heritable interindividual variation in platelet reactivity that may be relevant to these clinical events,3 and this heritability is greater in blacks than whites.4-6 However, there has been a paucity of literature considering racial differences in platelet function.
P2Y12 and thromboxane receptor inhibition with aspirin and thienopyridines are mainstays of antiplatelet therapy for arterial vascular disease. More recently, the protease-activated receptor (PAR)-1 antagonist, vorapaxar, was approved for the secondary treatment of patients with prior myocardial infarction and peripheral vascular disease. Although PAR4 antagonists have been developed,7-9 none of them have been studied in humans. Thrombin signals through platelet PAR1 and PAR4. These receptors couple to Gq proteins, leading to the activation of phospholipase Cβ, hydrolysis of phosphoinositides, and increased cytoplasmic calcium, resulting in activation of integrin αIIbβ3 and platelet aggregation.10 There are cellular phenotypic differences between PAR1 and PAR4.11-17 PAR1 has a higher affinity for thrombin, and Ca2+ transients rise sharply after PAR1 activation with PAR1-activating peptide (PAR1-AP), followed by a fast return to baseline levels. PAR4 stimulation with PAR4-activating peptide (PAR4-AP) induces a more gradual, but sustained, rise in [Ca2+]i, which accounts for the majority of intracellular calcium flux.12 PAR1 blockade with vorapaxar leaves PAR4 as the only means by which thrombin can activate platelets.18 PAR4 inhibition has a potential therapeutic advantage of inhibiting the maximal thrombin effect while minimizing bleeding because PAR1 signaling remains intact.19
We recently demonstrated 3.7-fold increased PAR4-mediated aggregation kinetics and greater calcium mobilization in platelets from black individuals compared with white individuals and noted that phosphatidylcholine transfer protein partially accounted for this racial difference.20 Our analyses also indicated a genomic contribution to platelet function that differs by race. Phosphatidylcholine transfer protein expression differences accounted for 18% of the racial difference in thrombin-induced PAR4-mediated platelet reactivity. We now report racially dimorphic single nucleotide polymorphisms (SNPs) in the PAR4 gene, F2RL3, that regulate PAR4 function and the inhibitory capacity of the PAR4 antagonist, YD-3. These findings have potential clinical significance because greater platelet-mediated thrombosis could contribute to worse outcomes in blacks than whites after coronary events, and the clinical risks and benefits of vorapaxar and other PAR antagonists by race are unknown.
Methods
The platelet RNA and expression-1 study and genotyping
As described earlier,20,21 154 healthy individuals (70 black, 84 white) were recruited between 2010 and 2011. Participant race/ethnicity was initially classified by self-identification but was subsequently validated by principal component analysis of 4.3 million genotypes from all 154 platelet RNA and expression-1 (PRAX1) participants.20 Additional healthy, male and female participants who self-identified as black or white were recruited in Philadelphia, Pennsylvania (cohort 2), for platelet calcium and PAR antagonist studies. These participants were genotyped for rs773902 or rs2227346, using TaqMan SNP Genotyping Assays (Life Technologies, Carlsbad, CA). Written informed consent was obtained from all participants with the approval of the institutional review boards of the Baylor College of Medicine in Houston, Texas, and Thomas Jefferson University in Philadelphia. Research was conducted in accordance with the Declaration of Helsinki.
PAR4 expression
mRNA levels from leukocyte-depleted platelets22 were profiled using the Affymetrix Human Gene 1.0ST array (Affymetrix, Santa Clara, CA). Platelet protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and probed with α-PAR423 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies (sc-25778; Santa Cruz Biotechnology, Dallas, TX). PAR4 and GAPDH bands were quantified using Image Studio software (Li-Cor, Lincoln, NE). Results are presented as PAR4 intensity normalized to GAPDH intensity. Microarray data are available in the Gene Expression Omnibus (accession number GSE49921).
Platelet phenotyping
Platelet-rich plasma was obtained from each participant, and light transmission aggregometry was performed as described.20,21 We developed an integrated agonist response score (ARS) that allowed precise differentiation among participants with the same maximal aggregation. The score is the weighted average, determined by principal component analysis, of the max and slope of the aggregation curve, measured by light transmission aggregometry in response to 1 or more concentrations of agonist. The concentrations used were 0.5 mg/mL arachidonic acid (AA); 4 μM adenosine 5′-diphosphate; 500, 750, or 2000 ng/mL α-CD9 antibody (Abcam, Cambridge, UK); 10 or 20 ng/mL collagen-related peptide (synthesized at Baylor College of Medicine and crosslinked with glutaraldehyde); 1 or 2 μM PAR1-AP (GL Biochem, Shanghai, China); and 50 or 75 μM PAR4-AP (GL Biochem, Shanghai, China). This score correlated strongly with a standard assessment of maximal percentage aggregation, but inclusion of slope (a minor contributor to the overall score and highly related to maximal aggregation) allowed distinction among platelets from subjects with the same maximal aggregation value.20
Genotype, quantitative trait locus, and racial variation analyses
DNA from the buffy coats was hybridized to the HumanOmni5 array (Illumnia Inc., San Diego, CA). To evaluate associations between genotype markers and PAR4 ARS, we used a multiple regression framework that permits statistical adjustment to account for covariates potentially influencing the trait in addition to the genotype information that was our primary focus. We tested the association between genotype markers within 50 kb of the F2RL3 transcription start and stop site and the PAR4 ARS. To model an additive effect, we converted a genotype of each marker into integer values [0, 1, 2], representing the number of copies of the major allele. This value was introduced to the multiple regression model as an independent variable (predictor) after controlling for other covariates, including self-identified race, sex, and age. The dependent variable is the PAR4 ARS, resulting in the following linear model equation:
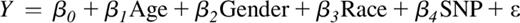
Here, Y represents the PAR4 ARS and ε is the stochastic error term. We estimated the β4 coefficient and its corresponding standard error, and we evaluated this model separately for each genotype marker. The P value for the β4 coefficient from this linear regression was used to assign significance between each genotype marker and PAR4 ARS.
Human Genome Diversity Project (HGDP) data and geographical representation were obtained, using the HGDP selection browser (http://hgdp.uchicago.edu/cgi-bin/gbrowse/HGDP/) from the Pritchard laboratory (University of Chicago, IL). The underlying data were generated by Li et al.24
Calcium mobilization
Washed platelets were resuspended to a final concentration of 1.0 × 106 platelets/mL in Tyrode’s buffer (137 mM NaCI, 0.3 mM Na2HP04, 2 mM KCI, 12 mM NaHCO3, 5 mM HEPES at pH 7.3, 5 mM glucose, 0.35% bovine serum albumin) supplemented with 1 mM CaCl2. Platelets were incubated with the cell-permeable Ca2+-sensitive dye, Fluo-4 AM, for 10 minutes, stimulated with 50 μM PAR4-AP, and mean fluorescence intensity (MFI) was measured in real-time on a flow cytometer for 10 minutes to monitor the rise in free intracellular Ca2+. Data are reported as the fold change, comparing maximum MFI to baseline MFI measured before platelet stimulation.
Inositol 1,4,5-triphosphate quantification
The expression vectors pBJ-FLAG-PAR4-120A-296F, pBJ-FLAG-PAR4-120A-296V, and pBJ-FLAG-PAR4-120T-296V were generated by site-directed mutagenesis from pBJ-FLAG-PAR4-120T-296F25 and sequenced for verification. Next, 2.5 × 106 293 human embryonic kidney cells were transfected with 5 μg vector, using Lipofectamine 2000 (Life Technologies). Forty-eight hours after transfection, 10% of the cells were incubated for 20 minutes at 37°C with a fluorescein isothiocyanate-α-FLAG antibody (F1804; Sigma-Aldrich, St. Louis, MO). Surface expression was quantified using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). MFI was calculated using FlowJo software. The remaining 90% of cells were counted and then treated with 1 mM PAR4-AP or control. Samples were then lysed at 5, 30, or 600 seconds, and inositol 1,4,5-triphosphate (IP3) was quantified using the Inositol-1,4,5-Trisphosphate [3H] Radioreceptor Assay Kit (Perkin Elmer, Waltham, MA), following the manufacturer’s protocol. Results were normalized to cell number and surface PAR4 expression.
Antagonist studies
Platelets were preincubated with the indicated concentrations of vorapaxar26 (Axon Medchem, Reston, VA) or YD-3 [1-benzyl-3-(ethoxycarbonylphenyl)-indazole]7 (a gift from Craig Lindsley, Vanderbilt University, Nashville, Tennessee), as previously described.27 Aggregation was then quantified after stimulation with 5 μM PAR1-AP or 100 μM PAR4-AP.
Statistical analysis
Multiple linear regression analysis was performed including the PAR4 protein level and the risk SNPs as explanatory variables and PAR4 reactivity as the dependent variable. To determine the fraction of variance of PAR4 reactivity explained by PAR4 protein levels and F2RL3 genotype, PAR4 protein level was brought into the model first, followed by rs773902 genotype. To test for an interaction between rs773902 and YD-3, we performed a linear regression, using aggregation as the dependent variable and PAR4-AP concentration, YD3 concentration, and rs773902 genotype as dependent variables. All statistical analyses were implemented using the R statistical package,28 SPSS version 19 (IBM, Armonk, NY), or GraphPad Prism (GraphPad Software, La Jolla, CA). The linkage disequilibrium (LD) heat map was generated using the R packages LDheatmap and genetics. There was no substantive racial difference in the linkage analysis of the F2RL3 region.
Results
Racial difference in platelet PAR4 expression
Figure 1A shows greater PAR4 reactivity in platelets from 70 black participants compared with 84 white participants (P = 6.8 × 10−9) in the PRAX1 study. Regardless of race, there appeared to be 3 groups of PAR4-AP responders: high (>70%), intermediate (∼30% to ∼60%), and low (<20%). Such a finding would be consistent with an additive genetic effect, but first we considered whether quantitative differences in PAR4 protein expression might contribute to racial differences in platelet PAR4 reactivity. Using platelets from all 154 participants, we found that blacks had 9% higher mean F2RL3 mRNA levels (P = .043; Figure 1B) and 14% higher PAR4 protein levels (P = .0427; Figure 1C). Although these small racial differences in PAR4 expression did not seem adequate to explain the 3.7-fold faster platelet aggregation in blacks20 or the apparent 3 response groups, we considered whether expression levels correlated with PAR4 reactivity, but observed none (Figure 1D). In addition, multiple linear regression analysis showed that PAR4 protein level explained only 0.2% of the observed variation in PAR4 reactivity and that PAR4 protein was not explanatory for PAR4 reactivity (P = .415).

SNPs in F2RL3 associate with PAR4 reactivity. (A) PAR4 ARS values of PRAX1 subjects by self-identified race. P = 6.8 × 10−9, 2-tailed t test. (B) Microarray analysis of F2RL3 gene expression. Values are normalized and log2 transformed. P = 0.043, 2-tailed t test. (C) PAR4 protein levels normalized to GAPDH. P = .0427, 1-tailed t test. (D) Correlation analysis of platelet PAR4 reactivity with PAR4 protein expression level.
SNPs in F2RL3 associate with PAR4 reactivity. (A) PAR4 ARS values of PRAX1 subjects by self-identified race. P = 6.8 × 10−9, 2-tailed t test. (B) Microarray analysis of F2RL3 gene expression. Values are normalized and log2 transformed. P = 0.043, 2-tailed t test. (C) PAR4 protein levels normalized to GAPDH. P = .0427, 1-tailed t test. (D) Correlation analysis of platelet PAR4 reactivity with PAR4 protein expression level.
F2RL3 loci associated with PAR4 reactivity
We next considered whether a qualitative (or functional) difference in PAR4 might contribute to the racial difference in PAR4 reactivity. Quantitative trait locus analysis of the PRAX1 cohort identified 3 F2RL3 SNPs, rs773904, rs773903, and rs773902, associated with platelet PAR4 reactivity (P values of 1.26 × 10−14, 1.26 × 10−14, and 9.15 × 10−16, respectively) after accounting for race, age, and sex (Figure 2A). Linear regression analysis indicated no association of PAR4 protein level with rs773904 (P = .491), rs773903 (P = .491), or rs773902 (P = .489). rs773904 and rs773903 are intronic (Figure 2B), whereas rs773902 is located in the second exon and alters residue 120 in the second transmembrane domain. The “G” allele of rs773902 encodes alanine (Ala), and the “A” allele encodes threonine (Thr). Because rs773902 is nonsynonymous and in strong LD with the 2 intronic SNPs (Figure 2B), we focused our subsequent analyses on rs773902. After controlling for protein level, the SNP rs773902 explains 48% of variability in PAR4 reactivity (by analysis of variance [ANOVA] partial sum of squares).

Association of racially dimorphic PAR4 variants with platelet PAR4 function. (A) Manhattan plot showing association of SNPs in the F2RL3 gene with platelet PAR4-AP reactivity. Each circle represents a SNP. The circles for rs773903 and rs773904 overlap. Y-axis is –log10 of the P values for association, controlling for age, race, and sex; x-axis, chromosomal location. (B) Schematic of F2RL3 and LD plot of SNPs in F2RL3. Nonsynonymous SNPs coding changes are shown above the schematic. Red rs numbers indicate SNPs identified in quantitative trait locus analysis; blue rs number indicates a less common SNP observed only in black subjects. (C) Genotype and allele frequency of rs773902 in black and white PRAX1 subjects. (D) Worldwide allele frequency of rs773902 in HGDP data set.
Association of racially dimorphic PAR4 variants with platelet PAR4 function. (A) Manhattan plot showing association of SNPs in the F2RL3 gene with platelet PAR4-AP reactivity. Each circle represents a SNP. The circles for rs773903 and rs773904 overlap. Y-axis is –log10 of the P values for association, controlling for age, race, and sex; x-axis, chromosomal location. (B) Schematic of F2RL3 and LD plot of SNPs in F2RL3. Nonsynonymous SNPs coding changes are shown above the schematic. Red rs numbers indicate SNPs identified in quantitative trait locus analysis; blue rs number indicates a less common SNP observed only in black subjects. (C) Genotype and allele frequency of rs773902 in black and white PRAX1 subjects. (D) Worldwide allele frequency of rs773902 in HGDP data set.
The allelic frequency of rs773902 in PRAX1 differed between self-identified blacks and whites (P= 4.31 × 10−16, Fisher’s exact). The frequency of the G allele was 37% in blacks and 81% in whites (Figure 2C). Conversely, the frequency of the A allele was 63% in blacks but only 19% in whites. We queried the HGDP24 to assess whether US racial genotypes were similar to other geographic locations and found corresponding racial rs773902 allele frequencies: the A allele (most common in US blacks) was most prevalent in sub-Saharan Africa and Papua New Guinea; the G allele (most common in US whites) was more prevalent elsewhere (Figure 2D).
PAR4 Thr/Ala dimorphism at residue 120 and Phe/Val at residue 296 significantly associate with platelet PAR4 function
PAR4-mediated platelet aggregation significantly differed by rs773902 genotype in the whole cohort (P = 9.15 × 10−16) (Figure 3A) and within black and white subjects (P = 4.3 × 10−9 and P = 3.1 × 10−8, respectively) (Figure 3B). Platelets from participants homozygous for Thr120 (more common among blacks) achieved the highest average maximum aggregation in response to PAR4-AP, whereas platelets from participants homozygous for Ala120 (more common among whites) achieved the lowest average maximum aggregation among both races. Heterozygotes showed an intermediate phenotype. F2RL3 rs773902 genotype showed no effect on the platelet aggregation response to AA (P = .0614), adenosine 5′-diphosphate (P = .5656), collagen-related peptide (P = .1639), α-CD9 antibody (P = .6923), or PAR1-AP (P = .2807) (Figure 3C). Further analyses of AA-induced platelet aggregation data showed no difference between Ala/Ala homozygotes and Thr/Thr homozygotes (P = .6325, pairwise t test).
![Figure 3. PAR4 variants and platelet function. Platelet PAR4 reactivity by rs773902 genotype for (A) all participants and (B) each race individually. P = 9.15 × 10−16, association of PAR4 reactivity with rs773902 genotype by linear regression. (C) Platelet ARS of PRAX1 subjects for AA, adenosine 5′-diphosphate, collagen-related peptide, α-CD9 antibody (CD9), PAR1-AP (PAR1), and PAR4-AP (PAR4), segregated by rs773902 genotype. One-way ANOVA showed no association between rs773902 and any agonist except PAR4-AP (PAR4-AP [A] is shown again for ease of comparison). (D) Platelet calcium flux from subjects genotyped for rs773902 (cohort 2). Platelets were treated with 50 μM PAR4-AP. n = 10 GG; n = 5 AA/AG. P = .03 1-tailed t test. (E) Platelet PAR4 reactivity by rs773902 and rs2227346 genotypes among self-identified black subjects in PRAX1. P = 1.75x10−8, association of PAR4 reactivity with rs2227346 genotype partial F-test after controlling for rs773902 genotype. For all panels, the box represents the interquartile range, the horizontal line in box is the median, and the whiskers represent 1.5 times interquartile range.](/view-large/figure/7439951/3450f3.jpeg)
PAR4 variants and platelet function. Platelet PAR4 reactivity by rs773902 genotype for (A) all participants and (B) each race individually. P = 9.15 × 10−16, association of PAR4 reactivity with rs773902 genotype by linear regression. (C) Platelet ARS of PRAX1 subjects for AA, adenosine 5′-diphosphate, collagen-related peptide, α-CD9 antibody (CD9), PAR1-AP (PAR1), and PAR4-AP (PAR4), segregated by rs773902 genotype. One-way ANOVA showed no association between rs773902 and any agonist except PAR4-AP (PAR4-AP [A] is shown again for ease of comparison). (D) Platelet calcium flux from subjects genotyped for rs773902 (cohort 2). Platelets were treated with 50 μM PAR4-AP. n = 10 GG; n = 5 AA/AG. P = .03 1-tailed t test. (E) Platelet PAR4 reactivity by rs773902 and rs2227346 genotypes among self-identified black subjects in PRAX1. P = 1.75x10−8, association of PAR4 reactivity with rs2227346 genotype partial F-test after controlling for rs773902 genotype. For all panels, the box represents the interquartile range, the horizontal line in box is the median, and the whiskers represent 1.5 times interquartile range.
PAR4 variants and platelet function. Platelet PAR4 reactivity by rs773902 genotype for (A) all participants and (B) each race individually. P = 9.15 × 10−16, association of PAR4 reactivity with rs773902 genotype by linear regression. (C) Platelet ARS of PRAX1 subjects for AA, adenosine 5′-diphosphate, collagen-related peptide, α-CD9 antibody (CD9), PAR1-AP (PAR1), and PAR4-AP (PAR4), segregated by rs773902 genotype. One-way ANOVA showed no association between rs773902 and any agonist except PAR4-AP (PAR4-AP [A] is shown again for ease of comparison). (D) Platelet calcium flux from subjects genotyped for rs773902 (cohort 2). Platelets were treated with 50 μM PAR4-AP. n = 10 GG; n = 5 AA/AG. P = .03 1-tailed t test. (E) Platelet PAR4 reactivity by rs773902 and rs2227346 genotypes among self-identified black subjects in PRAX1. P = 1.75x10−8, association of PAR4 reactivity with rs2227346 genotype partial F-test after controlling for rs773902 genotype. For all panels, the box represents the interquartile range, the horizontal line in box is the median, and the whiskers represent 1.5 times interquartile range.
PAR4-mediated platelet activation results in Ca2+ release. Figure 3D shows that platelets from donors with either 1 or 2 copies of the PAR4-Thr120 variant underwent an approximately 2-fold higher level of Ca+2 flux compared with PAR4-Ala120 homozygotes in response to PAR4-AP stimulation (P = .03).
Additional analysis of the PRAX1 data identified a second nonsynonymous F2RL3 SNP, rs2227346, not linked to rs773904, rs773903 or rs773902 (Figure 2B). rs2227346 alters residue 296 in the sixth transmembrane domain of PAR4; the common “T” allele encodes phenylalanine (Phe), and the less common “G” allele encodes valine (Val). No alleles from whites harbored the rs2227346 G variant. Although the numbers of Val-positive subjects were small in PRAX1, the presence of Val296 was associated with dramatically lower PAR4-mediated platelet aggregation compared with PAR4-296Phe (P = 1.75 × 10−8) (Figure 3E).
The PAR4 Thr/Ala-120 and Phe/Val-296 dimorphisms alter PAR4 signaling
A proximal step in PAR4-induced platelet signal transduction upstream of calcium release is hydrolysis of phosphatidyl inositol 4,5-biphosphate to IP3 and diacylglycerol.29 To determine whether these PAR4 variants differ in their capacity to generate IP3, we generated FLAG-tagged expression constructs for each of the 4 possible PAR4 alleles. Transient transfection of each construct into 293 HEK cells demonstrated similar surface expression (1-way ANOVA; P = .6543) (Figure 4A and supplemental Figure 1A, available on the Blood Web site). Forty-eight hours after transfection, cells were stimulated with PAR4-AP, and IP3 was quantified. Figure 4B shows that PAR4-Thr120-Phe296 generated more IP3 than PAR4-Ala120-Phe296 (P = .01; 2-tailed t test at 30 seconds), and that Val296-containing variants showed less activity than either Phe296-containing variant. Absolute IP3 quantification in picomoles generated from each of the 3 independent experiments is shown in supplemental Figure 1B. There was no correlation between variant surface expression and IP3 generation (Pearson r = 0.3783; P = .2253).
![Figure 4. Functional differences in IP3 generation. (A) Representative flow cytometry tracings for surface FLAG-PAR4 expression. 120A-296V indicates the expression construct for PAR4-Ala120-Val296, 120A-296F for PAR4-Ala120-Phe296, 120T-296F for PAR4-Thr120-Phe296, and 120T-296V for PAR4-Thr120-Val296. Blue, α-FLAG; red, IgG control. (B) IP3 generation in 293 cells transfected with FLAG-PAR4 variants or control stimulated with 1 mM PAR4-AP. IP3 levels were normalized to cell number and PAR4 surface expression. For each independent experiment, all data points were calculated relative to the IP3 quantified in the PAR4-Thr120-Phe296 expressing cells after 30 seconds of treatment (ie, the maximum value measured in all experiments [percentage of maximum IP3 level]). n = 3. P value for 120T-296F vs 120A-296F at 30s = 0.01, 2-tailed t test.](/view-large/figure/7439956/3450f4.jpeg)
Functional differences in IP3 generation. (A) Representative flow cytometry tracings for surface FLAG-PAR4 expression. 120A-296V indicates the expression construct for PAR4-Ala120-Val296, 120A-296F for PAR4-Ala120-Phe296, 120T-296F for PAR4-Thr120-Phe296, and 120T-296V for PAR4-Thr120-Val296. Blue, α-FLAG; red, IgG control. (B) IP3 generation in 293 cells transfected with FLAG-PAR4 variants or control stimulated with 1 mM PAR4-AP. IP3 levels were normalized to cell number and PAR4 surface expression. For each independent experiment, all data points were calculated relative to the IP3 quantified in the PAR4-Thr120-Phe296 expressing cells after 30 seconds of treatment (ie, the maximum value measured in all experiments [percentage of maximum IP3 level]). n = 3. P value for 120T-296F vs 120A-296F at 30s = 0.01, 2-tailed t test.
Functional differences in IP3 generation. (A) Representative flow cytometry tracings for surface FLAG-PAR4 expression. 120A-296V indicates the expression construct for PAR4-Ala120-Val296, 120A-296F for PAR4-Ala120-Phe296, 120T-296F for PAR4-Thr120-Phe296, and 120T-296V for PAR4-Thr120-Val296. Blue, α-FLAG; red, IgG control. (B) IP3 generation in 293 cells transfected with FLAG-PAR4 variants or control stimulated with 1 mM PAR4-AP. IP3 levels were normalized to cell number and PAR4 surface expression. For each independent experiment, all data points were calculated relative to the IP3 quantified in the PAR4-Thr120-Phe296 expressing cells after 30 seconds of treatment (ie, the maximum value measured in all experiments [percentage of maximum IP3 level]). n = 3. P value for 120T-296F vs 120A-296F at 30s = 0.01, 2-tailed t test.
The PAR4-Thr120 variant is resistant to pharmacological inhibition by YD-3
Recently, there has been an increasing interest in the therapeutic potential of PAR antagonists as antithrombotic agents. We sought to determine whether the common Ala120Thr PAR4 variant might affect pharmacological inhibition of either PAR1 or PAR4. As expected, vorapaxar efficiently inhibited platelet aggregation through PAR1 (Figure 5A) but had no effect on PAR4-induced platelet aggregation, regardless of rs773902 genotype (Figure 5B). YD-3 is a synthetic, low-molecular-weight, nonpeptide indazole derivative that specifically blocks PAR4-induced platelet aggregation.7,30 We found a significant interaction between genotype and YD-3 antagonist on the PAR4-induced aggregation response (P = .02) (Figure 5C). Individuals homozygous for PAR4-Thr120 were resistant to up to 1 μM YD-3 inhibition, whereas platelets from PAR4-Ala120 homozygotes were inhibited by 0.5 μM YD-3; heterozygotes appeared to have an intermediate response.

PAR4 variants are differentially susceptible to pharmacological inhibition. (A-C) Platelets from donors genotyped for rs773902 were washed, incubated with the indicated concentrations of vorapaxar (VPX) (A-B) or YD-3 (C), and then stimulated with (A) 5 μM PAR1-AP or (B-C) 100 μM PAR4-AP. Aggregation was measured using light transmission aggregometry. n = 3 each genotype. Horizontal lines indicate mean. P = .02.
PAR4 variants are differentially susceptible to pharmacological inhibition. (A-C) Platelets from donors genotyped for rs773902 were washed, incubated with the indicated concentrations of vorapaxar (VPX) (A-B) or YD-3 (C), and then stimulated with (A) 5 μM PAR1-AP or (B-C) 100 μM PAR4-AP. Aggregation was measured using light transmission aggregometry. n = 3 each genotype. Horizontal lines indicate mean. P = .02.
Discussion
There is a racial disparity in CHD outcomes, and platelets from healthy black subjects are hyperreactive through the PAR4 thrombin receptor when compared with platelets from whites. In the current work, we investigated the genetic and molecular mechanisms that contribute to racial differences in human platelet function. The major findings were the identification of racial differences in the frequency of common alleles of the PAR4-encoding gene, F2RL3, with whites having a high frequency of the Ala120 variant and blacks having a high frequency of the Thr120 variant; an effect of the Ala120Thr on platelet PAR4 function, wherein the Thr120 allele was associated with greater platelet aggregation, accounted for 48% of the variance in human platelet PAR4 reactivity and induced greater signaling; and an effect of the Ala120Thr variant on the inhibitory capacity of the PAR4 antagonist, YD-3. These findings have important implications for risk, outcome, and management of CHD.
There is a pronounced variation of 0% to 100% in the platelet aggregation response to PAR4-AP in the PRAX1 cohort, with platelets from black subjects showing greater reactivity than platelets from whites. Phosphatidylcholine transfer protein was previously shown to account for a portion of this racial difference, but ∼82% of the variance remained unexplained. We found that platelets from blacks expressed an average of 14% more PAR4 protein than platelets from whites. Although a small change in a surface receptor expression could theoretically have a larger effect on platelet aggregation, several aspects of our data do not support this possibility. First, there was no correlation between protein levels and PAR4-mediated platelet reactivity. Second, regression analysis indicated the protein levels accounted for only 0.2% of the observed variation in PAR4 reactivity. Third, the distribution of protein levels (Figure 1C) did not resemble the distribution of PAR4 reactivity (Figure 1A). For these reasons and because the 3 groups of PAR4 reactivity (high, intermediate, and low) were consistent with the possibility of 3 genotypes, we considered a genetic basis for the racial difference in PAR4 reactivity.
Three SNPs within F2RL3 met genomewide significance for associations with PAR4-mediated platelet reactivity. These SNPs map within 501 base pairs of one another and are in very strong LD. Two of these 3 SNPs are intronic, and we have not pursued their functionality. In addition to rs773902, we searched public databases and found reports of 10 other missense SNPs in F2RL3. However, each of these has a minor allele frequency of less than 1%, making it implausible these could account for the association between rs773902 and platelet aggregation in our 154-subject PRAX1 study. Blacks in PRAX1 more commonly express the rs773902 Thr120 allele, whereas whites more commonly express the Ala120 allele. The similar allele frequencies by race between PRAX1 and the HGDP argue against a US-specific finding and suggest our data may apply more broadly to global populations.
Only 2 large genomewide studies have been performed that tested for associations with platelet function. The first (from the Framingham Heart Study, Framingham, MA and Johns Hopkins University School of Medicine, Baltimore, MD) did not use agonists that activate platelets through PAR1 or PAR4.31 The second study was PRAX1.20 We have been unable to identify prior genomewide association study “hits” in F2RL3 associated with clinical thrombotic disease, but most studies were underrepresented with black patients. The Thr120 allele in the white population is only 16%, and perhaps these studies were not powered to identify associations among relatively infrequent variants such as this. Muehlschlegel et al performed a candidate locus substudy of the Coronary Artery Bypass Grafting Genomics Program and reported an association between rs773857 and PAR4-AP-mediated platelet P-selectin expression. This SNP is in the CPAMD8 gene, ∼18 kb downstream of F2RL3, but has no linkage to the F2RL3 SNPs we have described here.32
The rs773902 genotype was strongly associated with PAR4-induced platelet aggregation and calcium release, but not with the platelet aggregation response to other agonists, further supporting a PAR4-specific effect. A second amino acid-changing variant was identified, but only in black subjects. Transfection studies showed that PAR4-Val296 traffics to the membrane but does not signal normally. Intriguingly, the valine for Phe substitution functioned as a hypomorphic allele, the effect of which was dominant over the Thr120/Thr120 or Ala120/Thr120 variants. Because of the low frequency of this allele, it is difficult to make firm conclusions about its effect in human platelets.
To assess the biochemical effects of amino acid changes on downstream signaling events, we used 293 cells that contain the appropriate components (the Gq pathway) and have been a standard in the platelet biology field.33-35 Using PAR4-AP-induced IP3 generation, an important PAR4 signaling molecule,11,36 overexpression of the PAR4 variants demonstrated differing signaling capacities. A potential limitation of these experiments is that 293 cell type-specific effects may not apply to platelets. Unfortunately, we determined that megakaryocyte-like cell lines (Meg-01 and HEL) express endogenous PAR4, prohibiting unambiguous interpretation of transfection data. Nevertheless, taken together, the simplest explanation for these genetic and cell biologic studies is that these functional variants alter platelet function.
The mechanism by which F2RL3 variants affect function is unclear, but our data raise a number of hypotheses. The greater Ca2+ flux in platelets and enhanced IP3 generation in 293 cells expressing PAR4-Thr120 suggests greater Gq-induced activation of phospholipase Cβ Kobilka et al have shown that in other 7-transmembrane G-protein coupled receptors, the transmembrane core of the protein undergoes conformational changes on activation, affecting G-protein interaction.37 In addition, a Thr (polar) substitution for Ala (nonpolar) at residue 120 may affect the interaction between the receptor and cholesterol, and possibly affect both dimerization and ligand affinity.38 Regarding the less common Phe296Val variant in transmembrane helix 6, this residue is predicted to lie in the ligand-binding pocket of G-protein coupled receptor structures, and a valine substitution is predicted to alter ligand-binding properties (http://www.gpcr.org/7tm/). Future crystal structure information for these variants may address these possibilities.
In May 2014, the US Food and Drug Administration approved the PAR1 antagonist vorapaxar for the prevention of recurrent cardiovascular events in patients with prior myocardial infarction or peripheral vascular disease. As expected, we found that vorapaxar efficiently inhibited human platelet aggregation through PAR1, regardless of PAR4 genotype. Perhaps more relevant to this manuscript, vorapaxar had no effect on PAR4-induced platelet aggregation, regardless of genotype. The implications of our data on the clinical benefit or harm of vorapaxar are 2-fold: patients with a more active PAR4-Thr120 variant (mostly blacks) may not be as protected as whites (mostly Ala120) against arterial thrombosis while taking vorapaxar, and patients containing the less active PAR4-Ala120 variant (mostly whites) may have increased bleeding while taking vorapaxar. In the absence of rigorous pharmacogenetic studies powered for the allele most common in blacks, it is not possible to address these possibilities.
PAR4 inhibition may be an attractive alternative to PAR1 blockade by reducing the prolonged effects of thrombin while retaining the transient signaling mediated by an intact PAR1 pathway. In addition to blocking PAR4-induced, but not PAR1-induced, platelet aggregation,7,30 YD-3 also blocks thrombin-induced platelet recruitment and smooth muscle cell proliferation, as well as neutrophil-induced platelet aggregation.7,39,40 Oral YD-3 also attenuates intimal thickening in a rat carotid balloon injury model.40 We are unaware of the prior assessment of a pharmacogenetic interaction between YD-3 (or any PAR antagonist) and rs773902 for PAR4-induced platelet aggregation. Our data demonstrate a potent interaction between the PAR4 Ala120Thr variant and YD-3 inhibition of PAR4-induced platelet aggregation. We hypothesize that YD-3 induces conformational changes in PAR4, and the extent of the conformational change is regulated by the amino acid at residue 120. Crystal structure data of the 2 receptor isoforms with YD-3 may help elucidate the basic molecular mechanism of this pharmacogenetic effect.
In summary, the F2RL3 variants described in this work contribute to a major fraction of the racial variance in human platelet PAR4 reactivity. The common PAR4 Ala120Thr variants lead to different downstream signaling events and pharmacogenetic interactions, and the rs773902 genotype may be more important than self-identified race for predicting risk and benefit in CVD. Last, and perhaps most important, these data suggest that YD-3 could benefit whites more than blacks and that PAR4 hyperreactivity in blacks may negate some of the vorapaxar benefit. These apparent racial disparities support a need to develop new PAR4-antagonists that effectively block the PAR4-Thr120 variant; such compounds may be especially beneficial to patients with African ancestry.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by grants from the National Institutes of Health National Heart, Lung, and Blood Institute (HL102482) and National Institute on Minority Health and Health Disparities (MD007880) and funding from the Cardeza Foundation for Hematologic Research.
Authorship
Contribution: P.F.B. and C.A.S. designed the overall study; L.C.E. and P.F.B. wrote the manuscript; L.C.E., R.T.-M., M.H., X.K., and P.F.B. designed experiments; L.M.S., C.R.L., B.E.T., R.T.-M., X.K., and L.M. conducted experiments; L.C.E., L.M.S., C.R.L., R.T.-M., M.H., E.S.C., C.A.S., S.C., and P.F.B. analyzed data; and M.N. developed critical reagents.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul F. Bray, Cardeza Foundation for Hematologic Research, Sidney Kimmel Medical College, 1020 Locust St, Suite 394, Philadelphia, PA 19107; e-mail: paul.bray@jefferson.edu.

