Key Points
Variation at 6p21.31, 6q23.3, 11q23.1, 16p11.2, and 20q13.12 influences risk for HL.
Genetic predisposition implicates germinal center dysfunction, disrupted T-cell function, and NF-κB activation in the pathogenesis of HL.

Abstract
To further our understanding of inherited susceptibility to Hodgkin lymphoma (HL), we performed a meta-analysis of 7 genome-wide association studies totaling 5325 HL cases and 22 423 control patients. We identify 5 new HL risk loci at 6p21.31 (rs649775; P = 2.11 × 10−10), 6q23.3 (rs1002658; P = 2.97 × 10−8), 11q23.1 (rs7111520; P = 1.44 × 10−11), 16p11.2 (rs6565176; P = 4.00 × 10−8), and 20q13.12 (rs2425752; P = 2.01 × 10−8). Integration of gene expression, histone modification, and in situ promoter capture Hi-C data at the 5 new and 13 known risk loci implicates dysfunction of the germinal center reaction, disrupted T-cell differentiation and function, and constitutive NF-κB activation as mechanisms of predisposition. These data provide further insights into the genetic susceptibility and biology of HL.
Introduction
Hodgkin lymphoma (HL) comprises classical HL (cHL; ∼95% of cases) and nodular lymphocyte predominant HL (∼5% of cases).1 Although cHL and nodular lymphocyte-predominant HL are defined by the Hodgkin and Reed-Sternberg (HRS) cell and the lymphocyte-predominant cell, respectively, both diseases are thought to arise from the malignant transformation of germinal center B cell.2,3 Furthermore, both cHL and nodular lymphocyte-predominant HL demonstrate a paucity of these neoplastic B cells within a background of reactive inflammatory cells that includes large populations of CD4+ T cells.4,5
A viral or infectious agent has long been considered a major etiological factor for HL, with Epstein-Barr virus (EBV) being the posited infectious agent.6,7 However, the EBV genome is only identifiable in a variable number of HL cases, and epidemiological data support a causal role for the virus in EBV-positive HL only.8 Evidence for genetic susceptibility to HL is provided by the elevated familial risk, as well as the high concordance between monozygotic twins.9,10 More recently, genome-wide association studies (GWAS) have confirmed an HLA association for HL and have identified single nucleotide polymorphisms (SNPs) at 13 non-HLA loci influencing risk.11,12
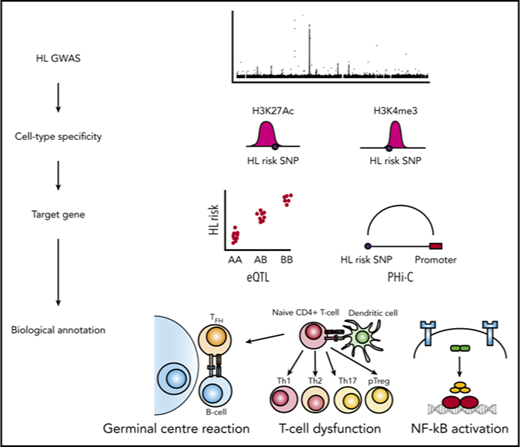
To gain further insight into HL susceptibility, we have conducted a meta-analysis of data from 7 independent GWAS and report 5 new HL risk loci.11-13 Integration of gene expression, histone modification, and in situ promoter capture Hi-C data (PCHi-C) at the 5 new and the 13 known risk loci provides evidence for cell-type specificity in B and T cells and implicates dysfunction of the germinal center reaction, disrupted T-cell differentiation and function, and constitutive NF-κB activation as mechanisms by which loci influence HL risk.
Materials and methods
Ethics
Collection of patient samples and associated clinicopathological information was undertaken with written informed consent. Relevant ethical review boards approved the individual studies in accordance with the tenets of the Declaration of Helsinki (UK-GWAS MREC 03/1/096, German-GWAS University of Heidelberg 104/2004, and UK-GWAS-NSHLG MREC 09/MRE00/72). The diagnosis of HL in all cases was established in accordance with World Health Organization guidelines.
GWAS
We used GWAS data generated on 3 nonoverlapping case–control series of Northern European ancestry, which have been the subject of previous analyses (supplemental Tables 1 and 2, available on the Blood Web site).11 The UK-GWAS was based on 622 cases ascertained through the Royal Marsden Hospital National Health Service Trust Family History study during 2004 to 2008,14 and 5677 control patients from the UK Wellcome Trust Case Control Consortium 2.15 The German-GWAS comprised 1001 cases ascertained by the German Hodgkin Study Group during 1998 to 2007, and 2092 control patients from the Heinz Nixdorf Recall study.16 The UK-NSHLG-GWAS used 1717 cases ascertained through the National Study of Hodgkin Lymphoma Genetics (https://www.ukctg.nihr.ac.uk) from 2010 to 2013.11 Control patients comprised 2976 cancer-free men recruited by the PRACTICAL Consortium, the UK Genetic Prostate Cancer Study (UKGPCS; age <65 years), a study conducted through the Royal Marsden NHS Foundation Trust and Study of Epidemiology & Risk Factors in Cancer, recruited via general practitioner practices in East Anglia (2003-2009); and 4446 cancer-free women from across the United Kingdom via the Breast Cancer Association Consortium. Details of the genotyping platform and quality control measures applied to each of the 3 GWAS have been described previously and are detailed in supplemental Tables 3 and 4.11,14,17,18 Briefly, individuals with a low call rate (<95%), as well as all individuals evaluated to be of non-European ancestry, were excluded (supplemental Figure 1). Eigenvectors for the GWAS data sets were inferred using smartpca (part of EIGENSOFT) by merging cases and controls with phase 3 HapMap samples.19 For apparent first-degree relative pairs, we excluded the control from a case–control pair or the individual with the lower call rate (supplemental Table 3). SNPs with a call rate below 95% were excluded, as were those with a minor allele frequency <0.01 or displaying deviation from Hardy–Weinberg equilibrium (ie, P < 10−6; supplemental Table 4). GWAS data were phased with SHAPEIT3,20 and imputed to more than 10 million SNP, using IMPUTE4 v1.021 and a merged reference panel consisting of data from 1000 Genomes Project (phase 3)22 and UK10K (EGAD00001000776).23 Imputation was conducted separately for each study from a set of SNPs common to cases and controls. Poorly imputed SNPs (defined by an information measure < 0.80) were excluded. Tests of association between SNPs and HL were performed using logistic regression under an additive genetic model in SNPTESTv2.5.2.24 The adequacy of the case–control matching was evaluated using Q–Q plots of test statistics (supplemental Figure 2). The inflation factor λ1000 was based on the 90% least-significant SNP scaled to 1000 cases and 1000 controls.
In addition to analyzing data from these 3 GWAS, we made use of preprocessed association test statistics for HL risk from a meta-analysis of 3 additional GWAS (USC-IARC-UC-GWAS) comprising 1816 HL cases and 7879 control individuals,12,25,26 and an analysis of 432 HL cases and 337 208 unaffected individuals13 from the UK Biobank, accessed through the Global Biobank Engine.
Meta-analysis
Meta-analyses were performed under a fixed-effects model, using META v1.7.24 Cochran’s Q-statistic to test for heterogeneity and the I2 statistic to quantify the proportion of the total variation resulting from heterogeneity were calculated; an I2 value at least 75% is considered to be characteristic of large heterogeneity.27 Where the same controls were used in both the UK-GWAS and the USC-IARC-UC GWAS, these controls were excluded from the UK-GWAS association analysis.
Cell culture
L-428 HL cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen and were cultured at 37°C in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific). Cell line identity was confirmed by STR-profiling. Cells were regularly tested for mycoplasma contamination (PromoCell, PK-CA91).
Chromatin immunoprecipitation-seq analysis
L-428.
Chromatin immunoprecipitation (ChIP)-seq was performed on H3K27Ac and H3K4me3 for using antibodies obtained from Diagenode. Briefly, after cell lysing, sonication of nuclei was performed (UCD-300, BioRuptor) to obtain 150- to 500-bp fragments. ChIP reaction was performed on a Diagenode SX-8G IP-Star Compact using Diagenode automated Ideal Kit reagents (C01010011). Protein A beads were incubated for 10 hours with 3 to 6 μg antibody and 2 to 4 million sonicated cell lysate. ChIP samples were de-cross-linked at 65°C for 4 hours and subsequently treated for 30 minutes with RNAse Cocktail and proteinase K. DNA was then purified (MiniElute PCR purification kit, Qiagen), followed by library preparation according to manufacture (HTP Illumina library preparation kit, KAPA Biosystems). Fourteen cycles of polymerase chain reaction were performed, followed by size selection for 200- to 400-bp fragments and final library purification (GeneRead Size Selection kit, 301 Qiagen). ChIP libraries were sequenced using HiSeq 2000 (Illumina) with 100-bp single-ended reads. Generated raw reads were filtered for quality (Phred33 ≥ 30) and length (n ≥ 32), and adapter sequences were removed using Trimmomatic v0.2235. Reads passing filters were then aligned to the human reference (hg19), using BWA v0.6.1. Peak calls are obtained using MACS2 v 2.0.1.
Histone modification data from primary blood cells.
H3K27Ac and H3K4me3 data from more than 100 samples from more than 30 cell types from the Blueprint Epigenome Consortium were analyzed.28
Cell-specificity analysis
Overlap enrichment analysis of HL risk SNPs with H3K4me3 and H3K27Ac peaks was performed as described by Trynka et al.29 Briefly, we evaluated whether the HL risk SNPs and SNPs in linkage disequilibrium (LD; r2 > 0.8) with the sentinel SNP were enriched at H3K4me3 and H3K27Ac ChIP-seq peaks in blood cells and the HRS cell line L-428 by a permutation procedure with 105 iterations.
Promoter capture Hi-C
In situ Hi-C libraries for L-428 were prepared as previously described.30,31 Briefly, 25 million cells were fixed in 1% formaldehyde for 10 minutes. Cross-linked DNA was digested with HindIII (NEB, R0104), and chromatin ends were filled and marked with biotin-14-dATP (Thermo Fisher, 19524-016). The resulting blunted ended fragments were ligated at 16°C in the nucleus with T4 DNA ligase (NEB, M0202) to minimize random ligation. DNA purified after cross-linking was reversed by proteinase K (Ambion, AM2546) treatment. DNA was sheared by sonication (Covaris, M220), and 200- to 650-bp fragments were selected. Biotin tag DNA was pulled down with streptavidin beads and ligated with Illumina paired-end adapters (Illumina). Six cycles of polymerase chain reaction were performed to amplify libraries before capture. Promoter capture was based on 32 313 biotinylated 120-mer RNA baits (Agilent Technologies), targeting both ends of HindIII restriction fragments that overlap Ensembl promoters of protein-coding, noncoding, antisense, snRNA, miRNA, and snoRNA transcripts (supplemental Data). After library enrichment, a postcapture polymerase chain reaction amplification step was carried out using 6 amplification cycles. Hi-C and PCHi-C libraries were sequenced using HiSeq 2000 technology (Illumina). Reads were aligned to the GRCh37 build using Bowtie2 v2.2.6,32 and identification of valid di-tags was performed using HiCUP v0.5.9.33 To declare significant contacts, HiCUP output was processed using CHiCAGO v1.1.8.34 Data from 3 independent biological replicates were combined to define a definitive set of contacts. Publicly accessible PCHi-C data generated in B- and T-cell populations were downloaded from the Open Science Framework.35
Chromatin interactions relevant to HL risk loci were defined as contacts overlapping with HL risk SNPs and SNPs in LD (r2 > 0.8 with the sentinel SNP), with promoters within a 2-Mb window of the sentinel SNP, and with a score of at least 5.0.34 Plotting of HL association data and chromatin contacts was performed using visPIG.36
Expression quantitative trait loci analysis
An analysis of associations between the SNPs (r2 > 0.8) at each locus and tissue-specific changes in gene expression was performed using summary statistics from 3 publicly available resources: lymphoblastoid cell line (LCL) expression from the MuTHER (n = 825) consortium37 ; LCL expression from the GTEx consortium (n = 114)38 ; and CD4+ and CD8+ T cells from 313 individuals.39 Statistical significance was assigned after correcting for the number of probes at each locus (microarray) or the number of transcripts at each locus (RNA-seq) for each expression data set.
Genetic correlation with infection
To estimate the genetic correlation between specific infections and all HL, and nodular sclerosis HL (NSHL) and mixed cellularity HL (MCHL) subtypes,40 we used LD score regression. Summary statistics for self-reported infectious diseases from more than 200 000 participants in 23andMe included41 chickenpox, shingles, cold sores, mononucleosis, mumps, hepatitis B, plantar warts, positive tuberculosis test results, Streptococcus throat infection, scarlet fever, pneumonia, bacterial meningitis, yeast infections, urinary tract infections, tonsillectomy, childhood ear infections, myringotomy, measles, hepatitis A, rheumatic fever, common colds, rubella, and chronic sinus infection.
Mendelian randomization
We performed 2-sample Mendelian randomization using SNPs associated with specific infection-related traits (ie, P ≤ 5.0 × 10−8) as instrumental variables.41 We analyzed infection-related traits for which more than 2 SNPs had been shown to be associated with the specific infection (tonsillectomy, mumps infection, childhood ear infection, and yeast infections). To avoid colinearity between SNPs for each trait, we excluded SNPs that were correlated (ie, r2 ≥ 0.01) within each trait and only considered the SNPs with the strongest effect on the trait for use as instrumental variables. Where data on an instrumental variable was not present in the outcome trait, a proxy was used (r2 > 0.6). Details of the instrumental variables used are detailed in supplemental Data. For each SNP, we recovered the chromosome position, risk allele, association estimates (per allele log-odds ratio [OR]), and standard errors. The allele that was associated with increased risk for the exposure was considered the effect allele. The ORs of HL, NSHL, and MCHL per unit of standard deviation increment for each infection-related trait were estimated using the Mendelian randomization R package.42 Given that traits analyzed are binary outcomes, the maximum likelihood method was employed with the resulting causal effect estimate representing the odds for HL risk per unit increase in the log-OR for infection-related trait.
Results
Association analysis
We analyzed summary level GWAS data generated on HL cases and controls of European ancestry11 from 3 sources (supplemental Tables 1-4): 2 GWAS of UK cases and controls and 1 GWAS of German cases and controls, totaling 3077 cases and 14 546 control patients (Discovery GWAS)11 ; the Stanford Global Biobank Engine, an analysis of 432 HL cases from the UK Biobank13 ; and a meta-analysis of 3 published HL GWAS totaling 1816 HL cases and 7879 control patients (USC-IARC-UC-GWAS).12,25,26
In a meta-analysis of data from the 7 studies, we identified new genome-wide significant associations for HL (Figure 1; Table 1) at 6p21.31 (rs649775; P = 2.11 × 10−10, marking ITPR3-UQCC2-IP6K3), 6q23.3 (rs1002658; P = 2.97 × 10−8, marking OLIG3-TNFAIP3), 11q23.1 (rs7111520; P = 1.44 × 10−11, marking POU2AF1), 16p11.2 (rs6565176; P = 4.00 × 10−8, marking MAPK3-CORO1A), and 20q13.13 (rs2425752; P = 2.01 × 10−8, marking NCOA5-CD40). In addition, we identified a promising association at 1p13.2 (rs2476601; P = 4.20 × 10−7, marking PTPN22).
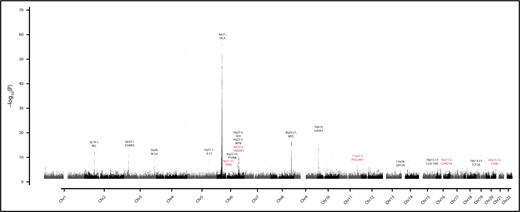
Genome-wide meta-analysis P values of Hodgkin’s lymphoma risk (–log10P, y-axis) plotted against their chromosomal positions (x-axis). Novel HL risk loci and candidate gene are in orange.
Genome-wide meta-analysis P values of Hodgkin’s lymphoma risk (–log10P, y-axis) plotted against their chromosomal positions (x-axis). Novel HL risk loci and candidate gene are in orange.
The bimodal incidence of HL and the higher rate of NSHL and EBV-negative HL in young adults suggest differences in the etiology of HL subtypes.8 Case-only analysis, however, provided no evidence for an age or histological subtype association for the 5 new-risk SNPs. (supplemental Tables 5 and 6).
Cell specificity of associations
Trynka et al have recently shown that chromatin marks highlighting regulatory regions overlap with phenotype-associated variants in a cell-type-specific manner.29 To examine for cell-type specificity of the 5 new and 13 known HL risk loci, we analyzed H3K4me3 and H3K27Ac chromatin marks, which annotate regulatory regions, in more than 125 samples from 38 hematopoietic cell types from BLUEPRINT28,29 and the HRS cell line L-428. The H3K27Ac histone mark is predominantly associated with enhancers, and of all the histone marks, it demonstrates the greatest enrichment of promoter-interacting regions.35 The H3K4me3 histone mark is predominantly associated with promoters and transcribed regions and has previously been shown to be the most phenotypically cell-type-specific.29,43 Cell types showing the strongest enrichment of risk SNPs at H3K4me3 marks were CD4+ T cells from venous blood (P = 2.9 × 10−3), CD3−CD4+CD8+-positive thymocytes (P = 5.7 × 10−3), and tonsillar-derived germinal center B cell (P = 6.3 × 10−3; supplemental Table 7). Cell types with the strongest enrichment of risk SNPs at H3K27Ac marks were CD8+ T cells from venous blood (P = 3.0 × 10−4), CD3+CD4+CD8+ thymocytes (P = 5.6 × 10−4), CD4+ thymocytes (P = 2.7 × 10−3), and L-428 (P = 7.9 × 10−3; supplemental Table 8). Based on the colocalization of variants with active chromatin marks, we calculated an enrichment score for each genetic association (Figure 2).29 High SNP regulatory scores were also shown in T cells at 3p24.1, 6q22.33, 6q23.3, and 10p14 risk loci; in B cells at 2p16.1, 3q28, 8q24.21, 11q23.1, and 20q13.12 risk loci; and in HRS cells at 3p24.1, 5q31.1, 6q22.33, 6q23.3, 10p14, 13q34 16p13.13, and 20q13.12.
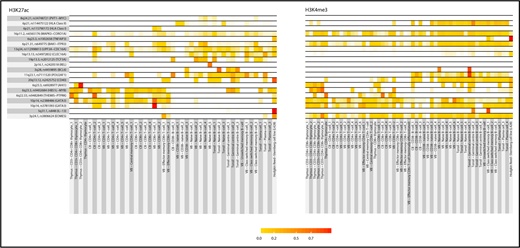
Heat map of SNP scores for H3K27Ac and H3K4me3 at each Hodgkin lymphoma risk locus. SNP score calculated as per Trynka et al.29 For each SNP at a given locus, the score represents the height of the closest ChIP-seq peak divided by the distance to the summit in each cell line, normalized across all immune cell types. Thus, a SNP within a chromatin mark that is active in only 1 cell type will have a high score of 1 (red) in that cell type and 0 (white) in others. In contrast, a SNP close to chromatin marks that are not cell-type-specific will have similarly modest scores across cell types. Genes at each risk locus are given for identification purposes only, and do not necessarily indicate biological functionality.
Heat map of SNP scores for H3K27Ac and H3K4me3 at each Hodgkin lymphoma risk locus. SNP score calculated as per Trynka et al.29 For each SNP at a given locus, the score represents the height of the closest ChIP-seq peak divided by the distance to the summit in each cell line, normalized across all immune cell types. Thus, a SNP within a chromatin mark that is active in only 1 cell type will have a high score of 1 (red) in that cell type and 0 (white) in others. In contrast, a SNP close to chromatin marks that are not cell-type-specific will have similarly modest scores across cell types. Genes at each risk locus are given for identification purposes only, and do not necessarily indicate biological functionality.
Identification of candidate target genes at HL risk loci
Most GWAS loci map to noncoding regions of the genome and influence gene regulation.44 Hence, to gain insight into the biological mechanisms for the associations at the 5 new and 13 known HL risk loci, we first performed expression quantitative trait locus (eQTL) analysis on expression data in B-cell LCLs and in CD4+ and CD8+ T cells. We identified eQTLs in LCLs at 6p21.31 (ITPR3), 6q23.3 (AHI1, ALDH8A1), 10p14 (GATA3), 11q23.1 (COLCA1, COLCA2), 13q34 (UPF3A, CDC16), 16p13.13 (SOCS1), 16p11.2 (MAPK3, BOLA2), and 20q13.12 (WFDC10B); in CD4+ T cells at 6q23.3 (AHI1) and 13q34 (CDC16); and in CD8+ T cell at 3p24.1 (EOMES), 6q23.3 (AHI1), and 13q34 (CDC16; Table 2; supplemental Tables 9 and 10).
Chromatin looping interactions between enhancer elements and promotors are central to regulation of gene expression.45 To link risk loci to candidate target genes, we analyzed PCHi-C data. First, we examined physical interactions at genomic regions annotated by HL risk loci (including variants with an r2 > 0.8), using publicly accessible PCHi-C in naive and total B cells, as well as CD4+ and CD8+ T cells.35 Second, we generated and analyzed PCHi-C data for the HRS cell line L-428. We observed concordance between H3K27Ac peaks and chromatin contacts in B, T, and HRS cells for specific HL risk loci. Notable chromatin contacts were found in the B lineage at 2p16.1 (REL), 6p21.31 (BAK1), 8q24.21 (MYC, PVT1), 13q34 (RASA3), 16p.13.13 (RMI2), and 20q13.12 (CD40); in the T-lineage at 3p24.1 (EOMES, AZI2), 6p21.31 (BAK1), 6q22.33 (THEMIS, PTPRK), 6q23.3 (MYB), 13q34 (RASA3), and 16p13.13 (SOCS1, RMI2); and in L-428 at 3p24.1 (AZI2, CMC1), 6q23.3 (MYB), 6q23.3 (TNFAIP3), and 16p13.13 (SOCS1, RMI2; Table 2; supplemental Figure 3; supplemental Data).
Shared susceptibility with infection
The association between EBV with HL, coupled with epidemiological reports of HL also being associated with non-EBV infections,46-49 suggests shared susceptibility a priori. Support for such an assertion is provided by a recent report implicating a number of the HL loci, including 6q23.3, 16p11.2, and 20q13.12, as well as the HLA region, as determinants of risk for infection.41
To investigate coheritability between HL and susceptibility to infection, we implemented cross-trait LD score regression.40 Using summary-level GWAS data, we estimated genetic correlations between HL and more than 20 self-reported infections in 200 000 23andMe participants.41 Overall, no statistically significant correlation was shown between any specific infection and HL, NSHL, or mixed cellularity HL (supplemental Table 11). Following on from this, for infections with greater than 2 genetically defined instrumental variables, we performed a Mendelian randomization analysis to identify a potential causal relationship with HL. For tonsillectomy, yeast infections, and childhood ear infections, no statistically significant associations were demonstrated (supplemental Table 12). A nominally significant positive association between self-reported mumps infection and HL was found (P = .04); however, this was not significant after correction for multiple testing.
Discussion
By using publicly available summary statistics, we have increased the power of our study, allowing us to identify 5 new HL risk loci, thus bringing the total number of HL risk loci to 18. Although our reliance on such data has restrained our ability to examine subtype-specific effects, it is likely that the newly described risk loci have generic effects on HL susceptibility, as with the known risk loci at 5q31.1 and 19p13.3.11
At the new and known HL risk loci, we observed an enrichment of active regulatory regions in germinal center B cells, CD4+ thymocytes, CD4+ T cells, and CD8+ T cells. Furthermore, although some HL risk loci locate to H3K27Ac peaks in both B and T cells, a number display lineage specificity. Motivated by this finding, we have used PCHi-C and gene expression data in these cell types to identify targets subject to regulatory control by HL risk SNPs. Although in part speculative, and requiring functional validation, integrating proximity, cell specificity of risk loci, gene expression, and PCHi-C data, our analyses highlight 3 biological processes and their associated genes as a basis of HL susceptibility (Table 2): the germinal center reaction (2p16.1, REL50 ; 3q28, BCL6 and mir-2851,52 ; 6p21, HLA53 ; 6q23.3, MYB54 ; 8q24.21, MYC55 ; 11q23.1, POU2AF156 ; 16p11.2, MAPK357 ; 19p13.3, TCF358 ; 20q13.12, CD40),59,60 T-cell differentiation and function (3p24.1, EOMES61 ; 5q31,1, IL1362 ; 6q22.33, PTPRK and THEMIS63,64 ; 6q23.3, MYB65 ; 6q23.3, AHI166 ; 10p14, GATA367 ; 16p13.1, SOCS1 and CLEC16A68,69 ; 16p11.2, MAPK3 and CORO1A70,71 ), and constitutive NF-κB activation (2p16.1, REL72 ; 3p24.1, AZI273 ; 6q23.3, TNFAIP374 ; 20q13.12, CD4075,76 ).
Our findings extend the relationship between germline genetics and tumor biology,44 as evidenced by enrichment of active chromatin marks for HL risk loci in L-428, and the findings of many of the target genes for HL GWAS associations are subject to somatic alterations in HRS cells; namely, REL,77 TNFAIP3, and SOCS1.78-80 The composite cellular basis of the HL tumor represents a preeminent example of the importance of the cellular microenvironment for the development of cancer. Hence, it is entirely conceivable that some of the HL risk loci may affect the development of the B-cell tumor indirectly. Support for such an assertion is the observation of T-cell specificity, as well as the finding of an eQTL at 3p24.1 (EOMES) in CD8+ T cells. Notably, EomesHi T-betLo PD-1HiCD8+ T cells are considered to delineate a key subset of exhausted CD8+ T cells,81,82 which may contribute to an immunosuppressive tumor microenvironment and is a feature of peripheral blood T cells in HL.83
There are a number of reasons for the observed lack of concordance between the PCHi-C and eQTL analysis at risk loci. First, the resolution of the Hi-C library using HindIII, a 6-base pair cutter, is approximately 10 kb. As such, we are unable to detect concordant chromatin contacts at risk loci, which influence the expression of genes located less than 10 kb. Second, it is recognized that the range at which gene expression is perturbed to influence disease risk may be narrow, and as such, it may not be detected by an eQTL analysis. Finally, given the risk loci are likely to act in specific cell populations, and our expression data are limited by broad B- and T-cell populations, it is possible that we have not captured the cell type to analyze expression. As such, we would view both methods as complimentary in identifying target genes.
The established association between EBV and risk for HL, coupled with other epidemiological observations, provides strong a priori evidence for infection being a major etiological risk factor for HL. Although our Mendelian randomization analysis did not implicate a causal relationship with any of the self-reported infection traits, we acknowledge that our study had limited power. It is, however, possible that pleiotropism between the 6p21.1, 6q23.3, 16p11.2, and 20q13.12 risk loci for HL and tonsillectomy is consistent with some form of a shared biological basis. This is intriguing, as tonsillectomy has previously been linked to HL in some epidemiological observational studies.46
In summary, our study provides further evidence for inherited susceptibility to HL and support for cell-type specificity at HL risk loci. Furthermore, through the integration of gene expression, histone modification and in situ PCHi-C data, our data highlight dysfunction of the germinal center reaction, perturbed T-cell function, and constitutive NF-κB activation as mechanisms by which genetic risk loci influence HL pathogenesis.
Sequencing data, which forms the reference panel for imputation, have been deposited in the European Genome-Phenome Archive under accession codes EGAD00001000776.
Transcriptional profiling data from the MuTHER consortium that support the findings of this work have been deposited in the European Bioinformatics Institute (Part of the European Molecular Biology Laboratory) under accession code E-TABM-1140.
ChIP-seq data from the Blueprint Epigenome Consortium are available from http://dcc.blueprint-epigenome.eu/#/home.
Hi-C data from the Blueprint Epigenome Consortium are available from https://osf.io/u8tzp/.
ChIP-seq data for the HRS cell line L-428 are deposited under the accession number EGAS00001003033.
Hi-C data for the HRS cell line L-428 are deposited in the European Genome-Phenome Archive under accession number EGAS00001003032.
Summary statistics for genetic susceptibility to infection-related traits are available upon request from 23andMe. Please visit research/23andme.com/collaborate to request access to these datasets.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. Patients for the new GWAS were recruited through the National Study of Hodgkin Lymphoma Genetics (https://www.ukctg.nihr.ac.uk). For their help with UK sample collection, the authors thank Hayley Evans, James Griffin, Joanne Micic, Susan Blackmore, Beverley Smith, Deborah Hogben, Alison Butlin, Jill Wood, Margot Pelerin, Alison Hart, Katarzyna Tomczyk, and Sarah Chilcott-Burns. The Breast Cancer Association Consortium study would not have been possible without the contributions of the following: Manjeet K. Bolla, Qin Wang, Kyriaki Michailidou, and Joe Dennis. For the British Breast Cancer study, the authors thank Eileen Williams, Elaine Ryder-Mills, and Kara Sargus. The authors thank the participants and the investigators of EPIC (the European Prospective Investigation into Cancer and Nutrition). The authors thank the Study of Epidemiology & Risk Factors in Cancer and EPIC teams. The authors thank Breast Cancer Now and the Institute of Cancer Research for support and funding of the UK Breakthrough Generations Study, and the study participants, study staff, and doctors, nurses, and other healthcare providers and health information sources who have contributed to the study. The UKGPCS would like to thank the Institute of Cancer Research and the Everyman Campaign for funding support. The UKGPCS acknowledges the Prostate Cancer Research Foundation, Prostate Action, the Orchid Cancer Appeal, the National Cancer Research Network UK, the National Cancer Research Institute, the National Institutes of Health Research funding to the National Institutes of Health Research Biomedical Research data managers and consultants for their work in the UKGPCS study, and urologists and other persons involved in the planning and data collection of the Cancer of the Prostate in Sweden study. The APBC BioResource, which forms part of the PRACTICAL consortium, consists of the following members: Wayne Tilley, Gail Risbridger, Renea Taylor, Judith A Clements, Lisa Horvath, Vanessa Hayes, Lisa Butler, Trina Yeadon, Allison Eckert, Pamela Saunders, Anne-Maree Haynes, and Melissa Papargiris. This study makes use of data generated by the Blueprint Consortium. A full list of the investigators who contributed to the generation of the data is available from www.blueprint-epigenome.eu. The authors thank Jose Martin-Subero (Institut d'investigacions Biomèdiques August Pi i Sunyer) for his advice with respect to the analysis of ChIP-seq data from the Blueprint Consortium. The authors also thank 23andMe for providing us with summary-level statistics of genetic associations to infection-related traits. Finally, the authors are grateful to all the patients and individuals for their participation, and the clinicians, investigators, other staff who contributed to sample and data collection.
The authors acknowledge use of genotype data from the British 1958 Birth Cohort DNA collection (funded by Medical Research Council grant G0000934 and Wellcome Trust grant 068545/Z/02) and the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant 090532/Z/09/Z). Funding for this project was provided by the Wellcome Trust under awards 076113 and 085475. The Breast Cancer Association Consortium is funded by Cancer Research UK (C1287/A10118, C1287/A16563). The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges National Health Service funding to the National Institutes of Health Research Biomedical Research Centre and the National Cancer Research Network. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Ligue Contre le Cancer; Institut Gustave Roussy; Mutuelle Générale de l’Education Nationale; Institut National de la Santé et de la Recherche Médicale; German Cancer Aid; German Cancer Research Center; Federal Ministry of Education and Research; the Hellenic Health Foundation; the Stavros Niarchos Foundation; Associazione Italiana per la Ricerca sul Cancro; National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports; Netherlands Cancer Registry; LK Research Funds; Dutch Prevention Funds; Zorg Onderzoek Nederland; World Cancer Research Fund; Statistics Netherlands; Health Research Fund; PI13/00061 to Granada; PI13/01162 to EPIC-Murcia; the regional governments of Andalucía, Asturias, Basque Country, Murcia, and Navarra; Instituto de Salud Carlos III Redes temáticas de investigación cooperativa en salud (RD06/0020); Cancer Research UK (14136 to EPIC-Norfolk, C570/A16491 and C8221/A19170 to EPIC-Oxford); and the Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford). The Study of Epidemiology & Risk Factors in Cancer and EPIC teams were funded by a program grant from Cancer Research UK (C490/A10124) and supported by the UK National Institutes of Health Research Biomedical Research Centre at the University of Cambridge. The authors acknowledge National Health Service funding to the Royal Marsden/ICR NIHR BRC. Genotyping of the OncoArray was funded by the National Institutes of Health (U19 CA 148537 for the Elucidating Loci Involved in Prostate Cancer Susceptibility project and X01HG007492 to the Center for Inherited Disease Research under contract number HHSN268201200008I). Additional analytic support was provided by the National Institutes of Health (NCI U01 CA188392). The PRACTICAL consortium was supported by Cancer Research UK Grants C5047/A7357, C1287/A10118, C1287/A16563, C5047/A3354, C5047/A10692, and C16913/A6135; European Commission's Seventh Framework Programme grant agreement no. 223175 (HEALTH-F2-2009-223175); and The National Institutes of Health Cancer Post-Cancer GWAS initiative grant: No. 1 U19 CA 148537-01 (the GAME-ON initiative). The authors would also like to thank the following for funding support: the Institute of Cancer Research and the Everyman Campaign, The Prostate Cancer Research Foundation, Prostate Research Campaign UK (now Prostate Action), the Orchid Cancer Appeal, the National Cancer Research Network UK, and the National Cancer Research Institute UK. The authors are grateful for the support of the National Institutes of Health Research funding to the National Institutes of Health Research Biomedical Research Centre at the Institute of Cancer Research and the Royal Marsden NHS Foundation Trust. Bloodwise (LLR, 10021) provided principal funding for the study. Support from Cancer Research UK (C1298/A8362) and the Lymphoma Research Trust is also acknowledged. A.S. is supported by a clinical fellowship from Cancer Research UK and charitable funds from the Royal Marsden Hospital. For the UK-GWAS, sample and data acquisition were supported by Breast Cancer Now, the European Union, and the Lymphoma Research Trust. The UK-GWAS made use of control genotyping data generated by the Wellcome Trust Case Control Consortium. Canadian Institutes of Health Research EP1-120608 supported the ChIP-seq work (T.P.). German funding was provided by the German Cancer Aid, the Harald Huppert Foundations, The German Federal Ministry of Education and Research (eMed, Cliommics 01ZX1309B), the Heinz Nixdorf Foundation, the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen, and the Faculty of Medicine University Duisburg-Essen. Funding for the project was provided by the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement no 282510 BLUEPRINT. M.M.N. has received funding for scientific projects from the Deutsche Forschungsgemeinschaft und the German Federal Ministry of Education and Science, fees for memberships in Scientific Advisory Boards from the Lundbeck Foundation and the Robert-Bosch-Stiftung and for membership in the Medical-Scientific Editorial Office of the Deutsches Ärzteblatt, and reimbursement for travel expenses for a conference participation by Shire Deutschland GmbH. M.M.N. also receives salary payments from Life & Brain GmbH and holds shares in Life & Brain GmbH.
Authorship
Contribution: A.S., K.H., and R.S.H. designed and provided overall project management; A.S. and R.S.H. drafted the manuscript; A.J.S., N.O., and R.S.H. provided samples for UK-GWAS and UK-NSHLG-GWAS; D.F.E., P.D.P.P., A.M.D., J.P., F.C., R.E., Z.K.-J., K.M., N.P., and D.C. provided control samples for the UK-NSHLG-GWAS; A.S. and P.J.L. performed bioinformatic and statistical analysis; A.S. and P.B. performed sample and laboratory coordination; A.S. and R.C. provided clinical data on the UK samples; A.S and G.O. performed capture Hi-C experiments; F.H. and T.P. performed ChIP-seq experiments; H.T. and A.F. performed bioinformatic and statistical analyses; P.H. and M.M.N. were responsible for German-GWAS analysis; K.-H.J. provided the German control samples; and E.P.v.S. and A.E. were responsible for German patients with HL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A list of the members of the PRACTICAL Consortium appears in “Appendix.”
Correspondence: Amit Sud, Division of Genetics and Epidemiology, The Institute of Cancer Research, London SW7 3RP, United Kingdom; e-mail: amit.sud@icr.ac.uk.
Appendix: study group members
The members of the PRACTICAL Consortium (http://practical.icr.ac.uk/) are: OncoArray (in addition to those named in the author list): Brian E. Henderson (University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, CA), Christopher A. Haiman (University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, CA), Sara Benlloch (The Institute of Cancer Research, London, United Kingdom/University of Cambridge, Cambridge, United Kingdom), Fredrick R. Schumacher (Case Western Reserve University, Cleveland, OH/Seidman Cancer Center, University Hospitals, Cleveland, OH), Ali Amin Al Olama (University of Cambridge, Cambridge, United Kingdom), Sonja I. Berndt (National Institutes of Health, Bethesda, MD), David V. Conti (University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, CA), Fredrik Wiklund (Karolinska Institute, Stockholm, Sweden), Stephen Chanock (National Institutes of Health, Bethesda, MD), Victoria L. Stevens (American Cancer Society, Atlanta, GA), Catherine M. Tangen (Fred Hutchinson Cancer Research Center, Seattle, WA), Jyotsna Batra (Queensland University of Technology/Translational Research Institute, Brisbane, Queensland, Australia), APCB BioResource (Queensland University of Technology, Brisbane, Queensland, Australia), Judith Clements (Queensland University of Technology/Translational Research Institute, Brisbane, Queensland, Australia), Henrik Gronberg (National Institutes of Health, Bethesda, MD), Johanna Schleutker (Translational Research Institute, Brisbane, Queensland, Australia/University of Turku/Turku University Hospital, Turku, Finland), Demetrius Albanes (University of Cambridge, Cambridge, United Kingdom), Stephanie Weinstein (University of Cambridge, Cambridge, United Kingdom), Alicja Wolk (Karolinska Institutet, Stockholm, Sweden), Catharine West (University of Manchester, Manchester, United Kingdom), Lorelei Mucci (Harvard School of Public Health, Boston, MA), Géraldine Cancel-Tassin (Tenon Hospital/Sorbonne Université, Paris, France), Stella Koutros (National Institutes of Health, Bethesda, MD), Karina Dalsgaard Sorensen (Aarhus University Hospital/Aarhus University, Aarhus C, Denmark), Eli Marie Grindedal (Oslo University Hospital, Oslo, Norway), David E. Neal (University of Cambridge/Addenbrooke’s Hospital/Cancer Research UK, Cambridge, United Kingdom), Ruth C. Travis (University of Oxford, Oxford, United Kingdom), Robert J. Hamilton (Princess Margaret Cancer Centre, Toronto, Canada), Sue Ann Ingles (University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, CA), Barry Rosenstein (Icahn School of Medicine at Mount Sinai, New York, NY), Yong-Jie Lu (Queen Mary University of London, London, United Kingdom), Graham G. Giles (The Cancer Council Victoria/The University of Melbourne, Melbourne, Victoria, Australia), Adam S. Kibel (Brigham and Womens Hospital, Boston, MA), Ana Vega (Fundación Pública Galega de Medicina Xenómica, Santiago de Compostela, Spain), Manolis Kogevinas (Barcelona Institute for Global Health, Barcelona, Spain/CIBER Epidemiología y Salud Pública, Madrid, Spain/Hospital del Mar Research Institute, Barcelona, Spain/Universitat Pompeu Fabra, Barcelona, Spain), Kathryn L. Penney (Brigham and Women’s Hospital/Harvard Medical School, Boston, MA), Jong Y. Park (Moffitt Cancer Center, Tampa, FL), Janet L. Stanford (Fred Hutchinson Cancer Research Center/University of Washington, Seattle, WA), Cezary Cybulski (Pomeranian Medical University, Szczecin, Poland), Børge G. Nordestgaard (University of Copenhagen, Copenhagen, Denmark/Copenhagen University Hospital, Herlev, Denmark), Hermann Brenner (German Cancer Research Center/National Center for Tumor Diseases, Heidelberg, Germany), Christiane Maier (University Hospital Ulm, Ulm, Germany), Jeri Kim (The University of Texas M. D. Anderson Cancer Center, Houston, TX), Esther M. John (Cancer Prevention Institute of California, Fremont, CA/Stanford University School of Medicine, Stanford, CA), Manuel R. Teixeira (Portuguese Oncology Institute of Porto/University of Porto, Porto, Portugal), Susan L. Neuhausen (Beckman Research Institute of the City of Hope, Duarte, CA), Kim De Ruyck (Ghent University, Ghent, Belgium), Azad Razack (University of Malaya, Kuala Lumpur, Malaysia), Lisa F. Newcomb (Fred Hutchinson Cancer Research Center/University of Washington, Seattle, WA), Davor Lessel (Medical University, Sofia, Bulgaria), Radka Kaneva (Medical University, Sofia, Bulgaria), Nawaid Usmani (University of Alberta/Cross Cancer Institute, Edmonton, Alberta, Canada), Frank Claessens (KU Leuven, Leuven, Belgium), Paul A. Townsend (University of Manchester/St Mary’s Hospital, Manchester, United Kingdom), Manuela Gago Dominguez (Servicio Galego de Saúde, Santiago De Compostela, Spain/University of California San Diego, La Jolla, CA), Monique J. Roobol (CE Rotterdam, the Netherlands), and Florence Menegaux (University Paris-Sud/University Paris-Saclay, Villejuif, France).