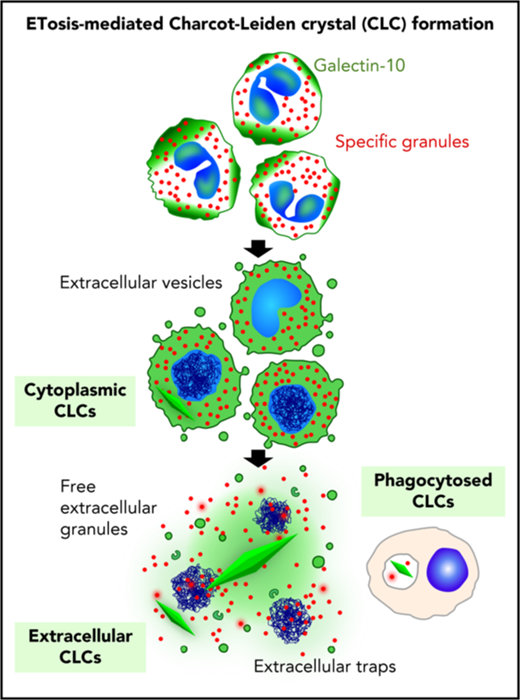
Key Points
This research reveals, for the first time, an active process by which CLCs are formed in eosinophilic diseases.

Abstract
Protein crystallization in human tissue rarely occurs. Charcot-Leyden crystals (CLCs) were described in various eosinophilic diseases >150 years ago, but our understanding of CLC formation still remains limited. In this study, we demonstrate that CLCs observed in varied inflamed human tissues are closely associated with eosinophil cell-free granules and nuclear envelope/plasma membrane disintegration with release of filamentous chromatin (extracellular traps), typical morphologies of a regulated pathway of extracellular trap cell death (ETosis). During the process of eosinophil ETosis, eccentrically localized cytoplasmic and perinuclear CLC protein (galectin-10) is homogeneously redistributed in the cytoplasm. Rapid (1-2 minutes) formation of intracytoplasmic CLCs was observed using time-lapse imaging. Plasma membrane rupture enabled the release of both intracellularly formed CLCs and soluble galectin-10 that further contributed to formation of CLCs extracellularly, in parallel with the expulsion of free intact granules and extracellular traps. CLC formation and galectin-10 release were dependent on nicotinamide adenine dinucleotide phosphate oxidase activation. To our knowledge, this is the first demonstration of natural formation of CLCs in association with an active physiological process (ie, ETosis). These results indicate that dynamic changes in intracellular localization and release of galectin-10 contribute to CLC formation in vivo and suggest that CLC/galectin-10 might serve as an indicator of ETosis.
Introduction
Cells need to transport proteins to their destination without premature crystallization.1 Protein-derived crystals seldom form spontaneously within human tissues, although Charcot-Leyden crystals (CLCs) are one of the exceptions.2 CLCs, slender bipyramidal hexagonal crystals found at sites of eosinophil infiltration in tissues, body fluids, and secretions, were identified >150 years ago and are associated with a variety of eosinophilic diseases, including asthma, myeloid leukemia, and allergic and parasitic diseases, and serve as hallmarks of active eosinophilic inflammation.3
CLC protein was initially identified as a lysophospholipase,4 but later assigned to the galectin superfamily, and named galectin-10.3 Human eosinophils contain a large pool of cytoplasmic CLC protein, constituting 7% to 10% of total cellular protein.3 Early studies demonstrated CLC formation through nonphysiological means in vitro by disrupting eosinophil-rich buffy coats with detergent or hypotonic treatments.4-6 Under such conditions, CLC formed within seconds or minutes, indicating a remarkable tendency of the free protein to form crystals. Studies identify the measurement of galectin-10 as a potentially useful biomarker of eosinophil involvement in eosinophil esophagitis,3,7 asthma,8 and allergic rhinitis.9 However, physiological mechanisms whereby CLCs are formed in vivo remained unknown.
Eosinophil cytolysis (also known as lytic degranulation) represents a regulated mode of cell death of activated eosinophils readily observed in vivo.10,11 Recent findings revealed that eosinophil cytolysis does not represent a process of accidental necrosis or apoptosis; rather, eosinophils are actively selecting their death program, namely extracellular trap cell death (ETosis).12 Eosinophil ETosis (EETosis) is a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase–dependent pathway culminating in nuclear envelope and plasma membrane disintegration, deposition of free extracellular granules (FEGs), and development of filamentous chromatin structures (extracellular traps, ETs) that promote inflammation and/or efficient elimination of pathogens. To date, EETosis has been evident in various allergic diseases, such as eosinophilic chronic rhinosinusitis (ECRS), eosinophilic otitis, and allergic bronchopulmonary aspergillosis.12-16 We reasoned that EETosis mediates CLC formation.
Study design
Written informed consent was obtained from all donors in accordance with the Declaration of Helsinki and under Institutional Review Board–approved protocols. Tissue specimens were obtained from patients with eosinophilic diseases, including ECRS.17 For in vitro studies, eosinophils were purified from normal donor blood by negative selection.12,13 Purity of isolated eosinophils was >98% of nucleated cells and viability was >99%. Biopsy samples and isolated cells were fixed and processed for transmission electron microscopy (TEM) as previously described.18 Specificities of immunostaining are shown in supplemental Figure 1, available on the Blood Web site. Other details are described in supplemental materials.
Results and discussion
First, we investigated CLCs in inflamed human tissues from varied eosinophilic diseases using TEM. As shown in Figure 1A-B and supplemental Figure 2, CLCs appeared as bipyramidal, hexagonal, and/or amorphous crystals in the interstitial tissues, adjacent to lytic eosinophils and closely associated with FEGs. Plasma membrane disintegration, chromatolysis, and/or loss of nuclear envelope, typical morphologies of EETosis, were frequently observed within the immediate vicinity of CLCs. Next, surgically obtained nasal polyps from patients with ECRS were assessed using hematoxylin and eosin staining. With careful observation by light microscopy, CLCs, acidophilic bipyramidal structures, were mainly located within clusters of eosinophils and/or free granules (Figure 1Ci). It has been previously shown that eosinophil cytolysis is correlated with disease severities.19 Overall among 36 patients (supplemental Table), CLCs were identified in sections obtained from 13 patients (36.1%), with the likelihood of CLC detection increasing in sections from patients with more severe disease (Figure 1Cii). Immunofluorescence staining for galectin-10 and an eosinophil granule protein (major basic protein, MBP) showed tissue migrating intact eosinophils with cytoplasmic/perinuclear-localized galectin-10 (Figure 1Di). In contrast, a highly inflamed eosinophilic lesion showed extracellular CLCs as well as small punctate galectin-10 staining within areas with extensive tissue depositions of MBP (Figure 1Dii). As previously described,20-22 small CLCs were occasionally observed within phagocytic macrophages and also within eosinophil cytoplasm (supplemental Figures 3-4).
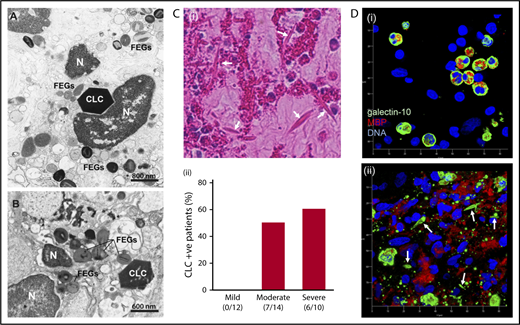
CLCs are associated with EETosis in human tissues. (A-B) Tissue CLCs in biopsies of frontal sinus (A, allergic patient) and bacterially infected colon (B, ulcerative colitis) with a large number of FEGs released by infiltrating lytic eosinophils. Note hexagonal crystals and chromatolytic nuclei (N). Samples were prepared for conventional TEM. (C) Evaluation of CLCs in nasal polyps from ECRS patients. (i) Arrows indicate typical CLCs (40× objective). Note the abundant eosinophils with chromatolysis and FEGs. (ii) The percentage of CLC-positive patients was assessed by hematoxylin and eosin staining according to clinical severities. Assessment of CLCs and detailed study subject information are provided in the supplemental materials. Numbers represent CLC-positive patients/total patients in each group. (D) Maximal projection of 3-dimensional z-stack images of galectin-10 (green) and MBP (red) staining of nasal polyps from ECRS patients. (i) A region of lesser eosinophil infiltration exhibiting intact eosinophils; (ii) a highly inflamed lesion with abundant CLCs. In panel Di, eosinophils with bilobed nuclei (blue) showed cytoplasmic/perinuclear galectin-10 staining. In contrast, small punctate galectin-10 and loss of cytoplasmic CLC with extracellular MBP were observed in panel Dii. CLCs with a bipyramidal structure (arrows) were stained with galectin-10. Images were obtained with a Carl Zeiss LSM780 confocal microscope (100× objective). The scale shows each 10 µm.
CLCs are associated with EETosis in human tissues. (A-B) Tissue CLCs in biopsies of frontal sinus (A, allergic patient) and bacterially infected colon (B, ulcerative colitis) with a large number of FEGs released by infiltrating lytic eosinophils. Note hexagonal crystals and chromatolytic nuclei (N). Samples were prepared for conventional TEM. (C) Evaluation of CLCs in nasal polyps from ECRS patients. (i) Arrows indicate typical CLCs (40× objective). Note the abundant eosinophils with chromatolysis and FEGs. (ii) The percentage of CLC-positive patients was assessed by hematoxylin and eosin staining according to clinical severities. Assessment of CLCs and detailed study subject information are provided in the supplemental materials. Numbers represent CLC-positive patients/total patients in each group. (D) Maximal projection of 3-dimensional z-stack images of galectin-10 (green) and MBP (red) staining of nasal polyps from ECRS patients. (i) A region of lesser eosinophil infiltration exhibiting intact eosinophils; (ii) a highly inflamed lesion with abundant CLCs. In panel Di, eosinophils with bilobed nuclei (blue) showed cytoplasmic/perinuclear galectin-10 staining. In contrast, small punctate galectin-10 and loss of cytoplasmic CLC with extracellular MBP were observed in panel Dii. CLCs with a bipyramidal structure (arrows) were stained with galectin-10. Images were obtained with a Carl Zeiss LSM780 confocal microscope (100× objective). The scale shows each 10 µm.
Various stimuli (interleukin -5/granulocyte-macrophage colony-stimulating factor with platelet activating factor, immobilized immunoglobulin G/immunoglobulin A, phorbol 12-myristate 13-acetate [PMA], and A23187) elicit NADPH-oxidase activation that leads to the common morphological process of eosinophil EETosis within 30 to 120 minutes in vitro.12,13 To test the hypothesis that EETosis mediates CLC formation, isolated human blood eosinophils were stimulated with known ETosis inducers and closely observed using time-lapse microscopy. Across all stimuli, CLCs were found in association with some cells within 30 minutes of stimulation. Time-lapse imaging and TEM showed that, during the loss of the eosinophil typical bilobed nucleus into a single round nucleus, nuclear shape change consistent with ETosis,12 CLC was rapidly (1-2 minutes) generated in the cytoplasm (Figure 2A; supplemental video 1; supplemental Figure 5). CLCs were rarely observed in either nonstimulated or EETosis inhibitor diphenyleneiodium chloride (DPI)-treated cells, indicating CLC formation is closely associated with an EETosis-mediated active process (Figure 2B; supplemental Figure 7A). To study the intracellular localization of galectin-10, stimulated cells were fixed at different time points and immunostained for galectin-10 (Figure 2C). Galectin-10 was initially distributed in the peripheral cytoplasm and perinucleus of adherent eosinophils. Over time, the eccentrically localized galectin-10 was homogeneously redistributed in the cytoplasm coincident with nuclear shape changes, followed by CLC formation in some cells. Subsequently, plasma membrane dissolution was accompanied by the appearance of extracellular CLC with FEGs and ETs (Figure 2Ciii; supplemental Figure 6). Time-lapse fluorescence images showed temporal immediacy of CLC formation coincident with compromised plasma membrane/nuclear envelope integrity (supplemental video 2) similar to that observed in electron micrographs from patient biopsies (Figure 1; supplemental Figure 2). During ETosis, eosinophils release plasma membrane–enveloped (but annexin V negative) extracellular vesicles (EVs).12 These EVs retained galectin-10 even after rupture of the originating cells (supplemental Figure 7B). The EVs, as well as released secretory vesicles (supplemental Figure 2C), likely correspond to the small punctate staining associated with CLCs in human tissues (Figure 1Dii).
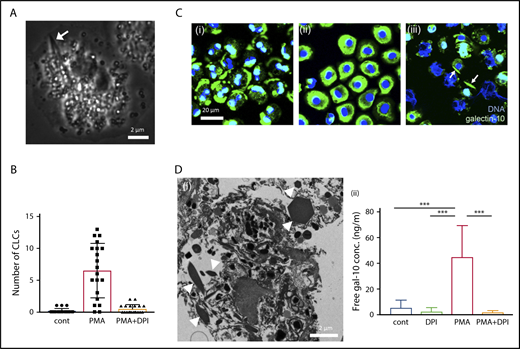
CLC formation in EETosis in vitro. (A) Cytoplasmic CLC formation in an eosinophil undergoing ETosis (arrow). Eosinophils were observed with time-lapse, phase-contrast imaging following IL-5 and platelet activating factor stimulation. Images were captured from supplemental video 1. (B) CLCs were associated with EETosis. Eosinophils were stimulated with PMA (10 ng/mL) with/without DPI for 120 minutes. CLCs were counted using an inverted microscope (40× objective, Eclipse TE300, Nikon). A total of 240 fields from 4 independent donors were studied. The bar graph represents the mean ± standard deviation. (C) Cellular galectin-10 localization during EETosis. PMA stimulated cells were fixed at 15, 45, and 120 minutes and stained with anti-galectin-10 Ab (green). At 15 minutes (i), galectin-10 was eccentrically located in adherent eosinophils. Galectin-10 was homogeneously distributed in cytoplasm at 45 minutes (ii). Note the loss of the typical bilobed nuclear shape. At 120 minutes (iii), cytoplasmic galectin-10 had disappeared and CLCs (arrows) could be recognized. ETs (filamentous DNA in blue) were also evident. EVs (supplemental Figure 7B) were out of focus. (D) Extracellular CLC formation. (i) Stacked EETosis cells form varied sizes of CLCs. Eosinophils (5 × 106 cells in 1.5 mL) were stimulated with PMA (10 ng/mL) in round-bottomed microtubes for 3 hours, followed by fixation and processing for TEM. Sectioned CLCs of variable size (arrowheads) were observed. (ii) Free galectin-10 levels in culture supernatants. Eosinophils were stimulated with PMA with/without DPI for 3 hours, and culture supernatants were recovered by centrifugation at 10 000g for 10 minutes to remove vesicles, free granules, and CLCs. The graph represents the mean ± standard deviation from 6 different donors. ***P < .001. cont, nonstimulated control.
CLC formation in EETosis in vitro. (A) Cytoplasmic CLC formation in an eosinophil undergoing ETosis (arrow). Eosinophils were observed with time-lapse, phase-contrast imaging following IL-5 and platelet activating factor stimulation. Images were captured from supplemental video 1. (B) CLCs were associated with EETosis. Eosinophils were stimulated with PMA (10 ng/mL) with/without DPI for 120 minutes. CLCs were counted using an inverted microscope (40× objective, Eclipse TE300, Nikon). A total of 240 fields from 4 independent donors were studied. The bar graph represents the mean ± standard deviation. (C) Cellular galectin-10 localization during EETosis. PMA stimulated cells were fixed at 15, 45, and 120 minutes and stained with anti-galectin-10 Ab (green). At 15 minutes (i), galectin-10 was eccentrically located in adherent eosinophils. Galectin-10 was homogeneously distributed in cytoplasm at 45 minutes (ii). Note the loss of the typical bilobed nuclear shape. At 120 minutes (iii), cytoplasmic galectin-10 had disappeared and CLCs (arrows) could be recognized. ETs (filamentous DNA in blue) were also evident. EVs (supplemental Figure 7B) were out of focus. (D) Extracellular CLC formation. (i) Stacked EETosis cells form varied sizes of CLCs. Eosinophils (5 × 106 cells in 1.5 mL) were stimulated with PMA (10 ng/mL) in round-bottomed microtubes for 3 hours, followed by fixation and processing for TEM. Sectioned CLCs of variable size (arrowheads) were observed. (ii) Free galectin-10 levels in culture supernatants. Eosinophils were stimulated with PMA with/without DPI for 3 hours, and culture supernatants were recovered by centrifugation at 10 000g for 10 minutes to remove vesicles, free granules, and CLCs. The graph represents the mean ± standard deviation from 6 different donors. ***P < .001. cont, nonstimulated control.
Cytoplasmic CLCs generated in culture plate wells in vitro were ∼5 to 10 μm in length, in contrast to varied sizes observed in vivo. To study whether increased local concentrations of free galectin-10 might crystalize extracellularly and thereby contribute to varied and larger CLC sizes observed in vivo, EETosis was induced in eosinophils cultured in round-bottomed tubes to allow cell stacking. As expected, CLCs of varied sizes were observed in cultures of stacked eosinophils undergoing EETosis (Figure 2Di). A substantial amount of free galectin-10 was released into the culture medium through a DPI-inhibitable mechanism (Figure 2Dii), indicating that galectin-10 is released through active, NADPH-oxidase–dependent cytolytic EETosis. This is also a novel finding because the secretory mode of galectin-10 had not been recognized, unlike well-described eosinophil secretion of intragranular proteins (ie, exocytosis or piecemeal degranulation).23
In this study, we addressed the close association of CLC formation and EETosis, an active form of cytolysis. Mechanisms to account for CLC formation in human tissue are proposed in supplemental Figure 8. More than a simple by-product of eosinophilic inflammation, CLC might be actively produced by programmed eosinophil cell death. Detection of filamentous ETs in solid tissue section is often difficult due to severely restricted extracellular spaces.24 In clinical settings, the presence of CLC and/or increased local galectin-10 concentration might serve as an alternative indicator of extensive occurrences of EETosis. Although the functional relevance of extracellular CLC is unknown, other types of crystals were implicated in inflammation through triggering tissue injury and cell death.25 Further investigation will shed light on the pathophysiological role(s) of CLC.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Noriko Tan, Yuka Nakamura, Satomi Misawa, Matthew Richard, Atsushi Wada, and Shinsuke Chida for their outstanding technical assistance. They also thank Ann M. Dvorak for providing clinical samples.
This study was funded in part by a Research Grant on Allergic Disease and Immunology from the Japan Agency for Medical Research and Development (JP18ek0410026), Charitable Trust Laboratory Medicine Research Foundation of Japan, Japanese Society of Laboratory Medicine Fund for Promotion of Scientific Research, and JSPS KAKENHI 15KK0329, 16K08926 (S.U.), 17K09993 (M.T.), and 17K17611 (Y.K.); Conselho Nacional de Desenvolvimento Científico e Tecnológico (R.C.N.M., Brazil) e Fundação de Amparo à Pesquisa do Estado de Minas Gerais FAPEMIG CBB-APQ-03647-16 (R.C.N.M., Brazil), and the National Institutes of Health, National Institute of Allergy and Infectious Diseases grants R37AI020241 (P.F.W.) and R01AI121186 (L.A.S.).
Authorship
Contribution: S.U. designed and performed experiments and wrote the manuscript; T.T., H.S., K.H., and S.F. provided clinical samples and edited the manuscript; R.C.N.M. performed electron microscopic experiments and wrote the manuscript; M.F., Y.K., and M.T. performed in vitro and immunostaining experiments; Y.Y. contributed histological studies of ECRS; L.A.S. contributed scientific advice and edited the manuscript; and M.H. and P.F.W. supervised the research and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shigeharu Ueki, Department of General Internal Medicine and Clinical Laboratory Medicine, Akita University Graduate School of Medicine, 1-1-1, Hondo, Akita 010-8543, Japan; e-mail: shigeharu.ueki@gmail.com.