

Key Points
Notch signaling drives the pathogenesis of cGVHD in mouse models of bone marrow transplantation.
Blockade of individual Notch ligands and receptors can prevent or treat distinct forms of cGVHD in mice.

Abstract
Chronic graft-versus-host disease (cGVHD) is a major complication of allogeneic hematopoietic cell transplantation (allo-HCT) and remains an area of unmet clinical need with few treatment options available. Notch blockade prevents acute GVHD in multiple mouse models, but the impact of Notch signaling on cGVHD remains unknown. Using genetic and antibody-mediated strategies of Notch inhibition, we investigated the role of Notch signaling in complementary mouse cGVHD models that mimic several aspects of human cGVHD in search of candidate therapeutics. In the B10.D2→BALB/c model of sclerodermatous cGVHD, Delta-like ligand 4 (Dll4)–driven Notch signaling was essential for disease development. Antibody-mediated Dll4 inhibition conferred maximum benefits when pursued early in a preventative fashion, with anti-Dll1 enhancing early protection. Notch-deficient alloantigen-specific T cells showed no early defects in proliferation or helper polarization in vivo but subsequently exhibited markedly decreased cytokine secretion and enhanced accumulation of FoxP3+ regulatory T cells. In the B6→B10.BR major histocompatibility complex–mismatched model with multi–organ system cGVHD and prominent bronchiolitis obliterans (BO), but not skin manifestations, absence of Notch signaling in T cells provided long-lasting disease protection that was replicated by systemic targeting of Dll1, Dll4, or both Notch ligands, even during established disease. Notch inhibition decreased target organ damage and germinal center formation. Moreover, decreased BO-cGVHD was observed upon inactivation of Notch1 and/or Notch2 in T cells. Systemic targeting of Notch2 alone was safe and conferred therapeutic benefits. Altogether, Notch ligands and receptors regulate key pathogenic steps in cGVHD and emerge as novel druggable targets to prevent or treat different forms of cGVHD.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) remains the only curative therapeutic option for many malignant and nonmalignant hematological disorders. Increased use of allo-HCT has been facilitated by improved donor–recipient matching and posttransplant supportive care. However, graft-versus-host disease (GVHD) is a major limitation to more successful allo-HCT.1-6 In particular, chronic GVHD (cGVHD) underlies the majority of nonrelapse posttransplant mortality and lifelong morbidity.6 The high burden of cGVHD is related to insufficient prevention and limited availability of effective therapies. Direct T-cell–mediated tissue injury driving acute GVHD (aGVHD) contributes to cGVHD development, but emerging evidence supports a broader interplay of immune mechanisms and tissue responses in cGVHD.7-11 In addition, temporal distinctions between aGVHD and cGVHD have been invalidated in preclinical and clinical studies, as chronic disease pathogenesis can be unleashed early after transplant.11-15
Notch is a highly conserved ligand-receptor signaling system well recognized as a key developmental regulator.16 In addition, Notch has been increasingly scrutinized for its role controlling peripheral T-cell responses in various disease pathologies.17,18 We identified a critical role for Notch signaling in the pathogenesis of aGVHD using multiple mouse allo-HCT models.19-21 Genetic blockade of Notch signaling in T cells with dominant-negative Mastermind-like (DNMAML; a truncated version of Mastermind-like1 coactivator and potent pan-Notch inhibitor) led to dramatically decreased GVHD in major histocompatibility complex (MHC)–mismatched and minor histocompatibility antigen–mismatched allo-HCT models, without causing global immunosuppression.19,20 Notch-deprived T cells had impaired production of multiple cytokines but preserved proliferation and expansion in vivo, increased accumulation of FoxP3+ regulatory T cells (Tregs), and potent antileukemic activity. Peritransplant effects of Notch blockade were mediated by Notch1/2 receptors in T cells and Delta-like ligands 1 and 4 (Dll1 and Dll4, respectively) in the host.21 Short-term antibody-mediated neutralization of Dll1/Dll4 in the peritransplant period was sufficient to provide the therapeutic benefits seen with genetic T-cell Notch inhibition, without deleterious intestinal side effects seen upon systemic treatment with γ-secretase inhibitors or anti-Notch1 antibodies.21,22 Key sources of Dll1/Dll4 ligands were discovered in nonhematopoietic fibroblastic stromal cells, with Dll1/4 inactivation in this subset conferring full protection from aGVHD.22 We also identified broader effects of Notch signaling in T-cell alloimmunity, as Notch blockade allowed long-term organ survival after murine heterotopic allogeneic heart transplantation through effects on T cells and alloantibody-mediated chronic rejection.23 Finally, emerging human data suggest cooperation of Notch with B-cell receptor signaling in cGVHD.24 Thus, we hypothesized that Notch signaling plays a role in cGVHD pathogenesis and that targeting Notch could mitigate disease severity in this area of unmet clinical need.
To investigate the role of Notch in cGVHD, we studied complementary mouse models of systemic cGVHD with dominant sclerodermatous changes (Scl-cGVHD; minor alloantigen-mismatched B10.D2→BALB/c model)25,26 or bronchiolitis obliterans disease manifestations (BO-cGVHD; MHC-mismatched B6→B10.BR model).8,27 New therapeutic opportunities can be effectively identified through combined use of these preclinical models.9,14,28,29 In Scl-cGVHD, inhibition of Dll1/Dll4–mediated Notch signals provided maximum protection if used early after transplant in a preventative fashion. Dll4 blockade accounted for most benefits, with disease control accompanied by Treg expansion and lasting inhibition of donor T-cell cytokine production. In this model, Notch blockade directly regulated the effector functions of alloantigen-specific T cells,30 without affecting T-cell expansion posttransplant. In the BO-cGVHD model, pan-Notch inhibition or Notch1/2 inactivation in T cells prevented BO and limited aberrant B-cell responses that are critical for disease development.8 In addition, Dll1, Dll4, or combined Dll1/Dll4 blockade reversed established disease manifestations. Thus, our data identify a critical role for active Notch signaling through Dll1 and Dll4 ligands during the continuum of effector T-cell and T-B reactivity in cGVHD pathogenesis. This work delineates the potential utility of several Notch-pathway–blocking agents for some of the most challenging pleomorphic manifestations of this devastating disease.
Materials and methods
Mice
BALB/c (H-2d, CD90.2+), B10.D2-Thy1.2 (H-2d, CD90.2+), and B10.D2-Thy1.1 (H-2d, CD90.1+) mice were bred at the University of Michigan. ROSA26DNMAMLf mice (B6-DNMAML; H-2b, CD45.2+) containing a Cre-inducible cassette encoding the DNMAML-GFP pan-Notch inhibitor under the ROSA26 promoter31 were backcrossed to the B10.D2-Thy1.1 background with Cd4-Cre for >9 generations (B10.D2-DNMAML; H-2d, CD90.1+). Cd4-Cre+Notch1f/f, Cd4-Cre+Notch2f/f, and Cd4-Cre+Notch1f/fNotch2f/f C57BL/6 mice were described before.21 C57BL/6 (H-2b) and B10.BR (H-2k) mice from The Jackson Laboratory were housed at the University of Minnesota. All experiments followed National Institutes of Health guidelines and were approved by institutional animal use and care committees.
Hematopoietic cell transplantation and GVHD analysis
For the Scl-cGVHD model, mice were lethally irradiated (8-8.5 Gy, 137Cs) and reconstituted with 8-10 × 106 T-cell–depleted (TCD) B10.D2-Thy1.2 bone marrow (BM) or TCD BM supplemented with 12 × 106 B10.D2-Thy1.1 wild-type (WT) or B10.D2-DNMAML splenocytes (∼3 × 106 T cells). As Cd4-Cre expression in WT T cells did not affect the phenotype and function of alloreactive T cells (supplemental Figure 1, available on the Blood Web site), all DNMAML negative littermates were used as controls. T-cell depletion was performed as described.32 For BO-cGVHD, B10.BR recipients were conditioned with cyclophosphamide (120 mg/kg/day i.p., day −3/−2; Sigma Aldrich) and irradiation (8.3 Gy, day −1), prior to reconstitution with 107 B6 TCD BM cells, or TCD BM plus 74-80 × 103 splenic T cells. Systemic clinical GVHD was graded using an established system.33 Cutaneous GVHD was assessed as described previously.34 Blinded histopathological scoring and scoring of apoptotic index on hematoxylin and eosin sections were performed by an experienced pathologist (M.A.P.).35 Semiquantitative analysis of skin fibrosis was performed by measuring dermal thickness.36 Collagen deposition was quantified on Masson’s trichrome–stained lung sections as a ratio of blue over total stained area.8
Pulmonary function tests
Pulmonary function tests were performed as described previously.27 Briefly, Nembutal-anesthetized mice were intubated and ventilated using the Flexivent system (Scireq). Pulmonary resistance, elastance, and compliance were determined using Flexivent software (v5.1).
Systemic antibody-mediated Notch inhibition
Use of neutralizing monoclonal antibodies (mAbs) specific for individual Notch ligands and receptors has been described in aGVHD.21 Briefly, humanized immunoglobulin G1 (IgG1) mAbs specific for Dll1 or Dll437,38 or for the Notch1 (NRR1) or Notch2 negative regulatory region (NRR2) were injected intraperitoneally (i.p.; 5 mg/kg on days 0, 3, 7, and 10 posttransplant, or as indicated in the figure legends). An anti–herpes simplex virus gD human IgG1 antibody was used as isotype control. For delayed targeting in Scl-cGVHD, 4 injections (5 mg/kg) were given over 10 days. In BO-cGVHD, anti-Dll1/anti-Dll4 mAbs (5 mg/kg) were mouse IgG2a to minimize anti-human antibody responses and injected i.p. biweekly for 4 weeks, starting once disease was established (day 28). A mouse anti-ragweed IgG2 mAb was used as isotype control. The quality of each mAb batch was tested by assessing inhibition of Dll4/Notch1-dependent T-cell development and Dll1/Notch2-dependent marginal zone B cells.39-42
Flow cytometry
Spleen, cutaneous lymph nodes, mesenteric lymph nodes, and liver, skin, and lung were collected at designated time points. Single-cell suspensions were prepared through cell strainers. Minced livers were digested for 30 minutes in RPMI + 10% fetal bovine serum containing 0.12 mg/mL Liberase TL and 0.2 mg/mL DNase I (Roche). Leukocytes were isolated by density gradient centrifugation (Accuprep, Accurate Chemical). Skin leukocytes were isolated as described previously.43 For cytokine detection, cells were restimulated for 6 hours in 96-well round-bottom plates precoated with 2.5 μg/mL anti-CD3ε/anti-CD28 (BioLegend), with brefeldin A (BD) during the last 4 hours. Surface staining was performed following staining with viability dye (BioLegend). Intracellular staining was done following the manufacturer’s protocol (eBioscience). Antibodies, clones, and manufacturers are described in supplemental Table 1. Analysis was performed on an LSRFortessa (BD). Sorting was performed with FACSAria III. Data were analyzed using FlowJo (Tree Star).
Quantitative analysis of gene expression
RNA was isolated from sort-purified Vβ3+CD4+ T-cells using TRIzol (Invitrogen) and RNeasy Micro Kit (QIAGEN), reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad), and subjected to polymerase chain reaction (PCR) using primers for Hprt1, Dtx1, Tbx21, and Gata3, Absolute Blue QPCR Mix (Invitrogen), and Mastercycler realplex (Eppendorf). Primer sequences were from PrimerBank44 (Hprt1, 7305155a1; Dtx1, 11611467a1; Tbx21, 9507179a1; and Gata3, 6679951a1).
Statistical analysis
GraphPad Prism 7.03 was used for statistical analysis. Values are presented as mean ± standard error of the mean. Statistical differences were calculated using 1-way analysis of variance (ANOVA) with Tukey correction for multiple comparisons, Student t-test, or log-rank test, as appropriate. Only statistically significant differences with P < .05 are highlighted.
Results
Notch blockade in mature T cells prevents the development of sclerodermatous cGVHD
We previously demonstrated that Notch signaling in donor CD4+ and CD8+ T cells is critical to promote aGVHD in multiple mouse models.19-21 To evaluate the role of Notch in cGVHD, we relied on complementary models of cGVHD in mice: (1) the B10.D2→BALB/c minor histocompatibility antigen–mismatched allo-HCT model with sclerodermatous disease features, including skin, liver, and lung fibrosis (Scl-cGVHD)25 ; and (2) the B10.BR→C57BL/6 MHC-mismatched model with multi–organ system disease without skin but with prominent bronchiolitis obliterans (BO-cGVHD) manifestations.27 These models differ in systemic inflammation intensity as well as in kinetic and clinical disease features, thus modeling different aspects of human cGVHD.
To define the overall role of Notch signaling in donor T cells in Scl-cGVHD, we first used T cells expressing the pan-Notch inhibitor DNMAML in mature T cells, which inhibits transcriptional activation downstream of all Notch receptors during antigen-driven immune responses.31 Lethally irradiated BALB/c mice received TCD BM with or without a T-cell inoculum from B10.D2 controls (WT) or B10.D2-DNMAML donors (Cd4-Cre+ROSA26DNMAML mice, expressing DNMAML in all T cells). Following allo-HCT, animals were monitored for clinical GVHD signs using established scoring systems assessing aGVHD33 and cGVHD.34 While recipients of WT T cells developed severe GVHD, clinical GVHD scores were markedly decreased for both overall systemic and cutaneous features (reflective of aGVHD and cGVHD, respectively) in mice receiving DNMAML T cells (Figure 1A-C; P < .001, WT vs DNMAML). WT but not DNMAML recipients exhibited prominent clinical cGVHD findings (alopecia, skin ulcerations, and contractures) and histopathological cGVHD features (destruction of adnexal structures, fibrosis and increased dermal thickness) (Figure 1D-E; supplemental Figure 2A). In this model featuring concurrent aGVHD and cGVHD phenotypes, histopathological aGVHD findings in skin (dyskeratosis and epidermal apoptosis) and other organs were prominent in recipients of WT, but not DNMAML, T cells (supplemental Figure 2B). Thus, Notch inhibition in T cells prevented manifestations of aGVHD and cGVHD.
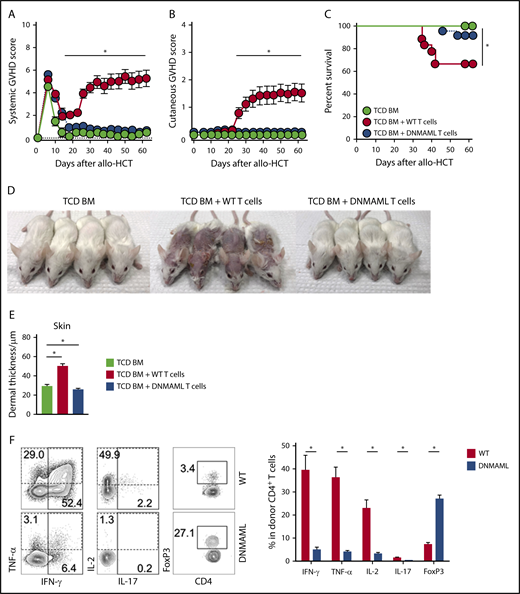
Notch blockade in mature T cells prevents sclerodermatous cGVHD. BALB/c mice were lethally irradiated (8-8.5 Gy), followed by transplantation of B10.D2-Thy1.2 TCD BM (light green circle), TCD BM with 12 × 106 B10.D2-Thy1.1 WT splenocytes (red circle), or 12 × 106 B10.D2-DNMAML splenocytes (blue circle), followed by monitoring with systemic or skin-specific clinical GVHD scores. B10.D2-DNMAML T cells cannot respond to canonical Notch signals. (A) Mean systemic clinical GVHD scores. *P < .01 (WT vs DNMAML T cells and WT T cells vs TCD BM only; 1-way ANOVA). (B) Mean skin-specific clinical GVHD scores; *P < .01 (WT vs DNMAML T cells and WT T cells vs TCD BM only; 1-way ANOVA). (C) Overall survival; *P < .001 (WT vs DNMAML T cells and WT T cells vs TCD BM only; log-rank test). n = 20 mice/group, pooled from 4 experiments. (D) Representative images of recipient mice on day 35 show absence of Scl-cGVHD features in TCD BM and DNMAML T-cell recipients. (E) Histopathological assessment of dermal thickness, a hallmark of skin cGVHD (n = 14, 3 experiments). *P < .01 (1-way ANOVA). (F) Flow cytometric assessment of inflammatory cytokine production and FoxP3 expression in donor-derived spleen T cells (day 6). Representative contour plots (left). Numbers indicate the percentage of events falling within indicated rectangular gates. Cumulative quantification (right) (n = 10 mice/group, from 3 experiments). *P < .001 (2-tailed unpaired Student t test). IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Notch blockade in mature T cells prevents sclerodermatous cGVHD. BALB/c mice were lethally irradiated (8-8.5 Gy), followed by transplantation of B10.D2-Thy1.2 TCD BM (light green circle), TCD BM with 12 × 106 B10.D2-Thy1.1 WT splenocytes (red circle), or 12 × 106 B10.D2-DNMAML splenocytes (blue circle), followed by monitoring with systemic or skin-specific clinical GVHD scores. B10.D2-DNMAML T cells cannot respond to canonical Notch signals. (A) Mean systemic clinical GVHD scores. *P < .01 (WT vs DNMAML T cells and WT T cells vs TCD BM only; 1-way ANOVA). (B) Mean skin-specific clinical GVHD scores; *P < .01 (WT vs DNMAML T cells and WT T cells vs TCD BM only; 1-way ANOVA). (C) Overall survival; *P < .001 (WT vs DNMAML T cells and WT T cells vs TCD BM only; log-rank test). n = 20 mice/group, pooled from 4 experiments. (D) Representative images of recipient mice on day 35 show absence of Scl-cGVHD features in TCD BM and DNMAML T-cell recipients. (E) Histopathological assessment of dermal thickness, a hallmark of skin cGVHD (n = 14, 3 experiments). *P < .01 (1-way ANOVA). (F) Flow cytometric assessment of inflammatory cytokine production and FoxP3 expression in donor-derived spleen T cells (day 6). Representative contour plots (left). Numbers indicate the percentage of events falling within indicated rectangular gates. Cumulative quantification (right) (n = 10 mice/group, from 3 experiments). *P < .001 (2-tailed unpaired Student t test). IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
To explore cellular mechanisms of GVHD protection, we assessed the impact of Notch blockade on early T-cell reconstitution and T helper responses. CD90.1+ graft-derived T cells were retrieved from secondary lymphoid organs and a representative GVHD target organ (liver) on day 6 after allo-HCT. This early time point revealed efficient Notch blockade and predicted aGVHD outcomes in multiple models.19-21 As seen in aGVHD,19,20 DNMAML expression did not inhibit allogeneic T-cell expansion in lymphoid organs and liver, but it decreased skin infiltration (supplemental Figure 2C-D), dramatically suppressed inflammatory cytokine production, and enhanced accumulation of Tregs (Figure 1F; supplemental Figure 2E-F). Taken together, these findings suggest that Notch blockade in donor T cells hinders early effector differentiation crucial for subsequent development of Scl-cGVHD in the B10.D2→BALB/c model.
Early Dll4-driven Notch signaling is key for induction of Scl-cGVHD in the B10.D2→BALB/c model
To identify elements of the Notch signaling cascade driving Scl-cGVHD development, we administered mAbs neutralizing specific individual Notch receptors or ligands.21,22 Lethally irradiated BALB/c mice received isotype control or designated mAbs starting immediately before transplant. Targeting Notch1 or Notch1/2 with anti-NRR1/2 neutralizing antibodies profoundly suppressed cytokine production and induced Treg expansion, similar to DNMAML-mediated pan-Notch inhibition in T cells (supplemental Figure 3A). Anti-NRR2 alone had limited effects on surrogate immunological end points and skin cGVHD manifestations (supplemental Figure 3A-C). However, systemic Notch1 blockade was poorly tolerated after transplantation due to intestinal side effects (not shown), as observed in aGVHD.21 In contrast, anti-Dll4 alone potently protected from GVHD without systemic side effects (Figure 2A-D; supplemental Figure 4A-C), with modest additional benefits from anti-Dll1. Flow cytometric analyses of allogeneic T cells replicated patterns of cytokine suppression, Treg accumulation, and long-term persistence seen with genetic pan-Notch blockade (supplemental Figure 4D-E). These data established the importance of Notch1 and Dll4-driven Notch signaling in Scl-cGVHD pathogenesis, with a safety profile favoring the targeting of Delta-like ligands over Notch1 receptor.
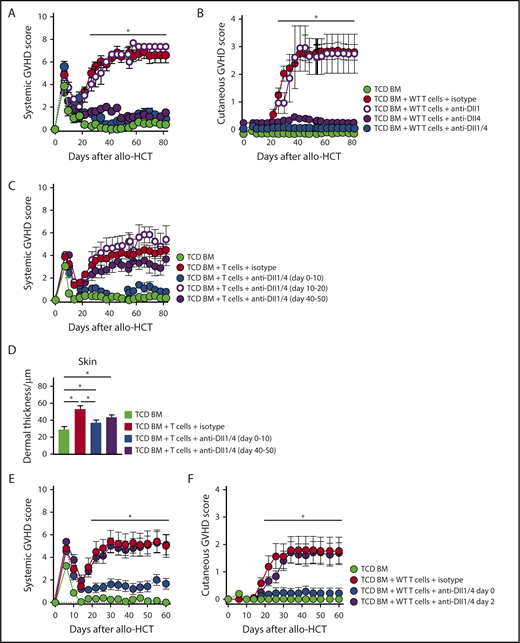
Dll1/Dll4 Notch ligands are critically active during the earliest posttransplant period and can be therapeutically targeted to prevent Scl-cGVHD. B10.D2→BALB/c chimeras were generated by transplanting B10.D2-Thy1.2 TCD BM (light green circle) vs TCD BM plus 12 × 106 B10.D2-Thy1.1 WT splenocytes. T-cell recipients were treated with 4 peritransplant doses of isotype control (red circle), anti-Dll1 (purple-rimmed circle), anti-Dll4 (purple circle), or anti-Dll1 plus anti-Dll4 (blue circle) antibodies (5 mg/kg i.p. on days 0, 3, 7, and 10). (A-B) Biweekly systemic and cutaneous GVHD scores, showing that Dll4 is the dominant Notch ligand in the Scl-cGVHD model and that its targeting induces almost complete disease protection. *P < .01 (1-way ANOVA) for TCD BM and all groups receiving anti-Dll4 vs groups receiving isotype control or anti-Dll1 alone (cumulative data from 2 experiments). (C-D) B10.D2→BALB/c mice were generated as described above. T-cell recipients were treated with 4 doses of isotype control (red circle) or anti-Dll1/4 (blue circle) antibodies at days 0 to 10 vs anti-Dll1/Dll4 at later time points (days 10-20, purple-rimmed circle; days 40-50, purple circle). (C) Systemic GVHD scores were monitored longitudinally. Note lack of GVHD protection with delayed administration of anti-Dll1/4. (D) Histopathological analysis of dermal thickness in mice depicted in panel C. *P < .01 (1-way ANOVA). Note Scl-cGVHD protection with early anti-Dll1/Dll4 administration and trend toward benefit with delayed anti-Dll1/Dll4 treatment (P = .07). (E-F) B10.D2→BALB/c chimeras were generated as described above. In one cohort, anti-Dll1/Dll4 treatment was delayed by 48 hours (purple circle). Note loss of protection from systemic (E) and cutaneous (F) GVHD with delayed anti-Dll1/4 administration. *P < .01 (1-way ANOVA) for TCD BM and group receiving early anti-Dll1/4 vs groups receiving isotype control or delayed anti-Dll1/Dll4 (n = 12, cumulative data from 3 experiments).
Dll1/Dll4 Notch ligands are critically active during the earliest posttransplant period and can be therapeutically targeted to prevent Scl-cGVHD. B10.D2→BALB/c chimeras were generated by transplanting B10.D2-Thy1.2 TCD BM (light green circle) vs TCD BM plus 12 × 106 B10.D2-Thy1.1 WT splenocytes. T-cell recipients were treated with 4 peritransplant doses of isotype control (red circle), anti-Dll1 (purple-rimmed circle), anti-Dll4 (purple circle), or anti-Dll1 plus anti-Dll4 (blue circle) antibodies (5 mg/kg i.p. on days 0, 3, 7, and 10). (A-B) Biweekly systemic and cutaneous GVHD scores, showing that Dll4 is the dominant Notch ligand in the Scl-cGVHD model and that its targeting induces almost complete disease protection. *P < .01 (1-way ANOVA) for TCD BM and all groups receiving anti-Dll4 vs groups receiving isotype control or anti-Dll1 alone (cumulative data from 2 experiments). (C-D) B10.D2→BALB/c mice were generated as described above. T-cell recipients were treated with 4 doses of isotype control (red circle) or anti-Dll1/4 (blue circle) antibodies at days 0 to 10 vs anti-Dll1/Dll4 at later time points (days 10-20, purple-rimmed circle; days 40-50, purple circle). (C) Systemic GVHD scores were monitored longitudinally. Note lack of GVHD protection with delayed administration of anti-Dll1/4. (D) Histopathological analysis of dermal thickness in mice depicted in panel C. *P < .01 (1-way ANOVA). Note Scl-cGVHD protection with early anti-Dll1/Dll4 administration and trend toward benefit with delayed anti-Dll1/Dll4 treatment (P = .07). (E-F) B10.D2→BALB/c chimeras were generated as described above. In one cohort, anti-Dll1/Dll4 treatment was delayed by 48 hours (purple circle). Note loss of protection from systemic (E) and cutaneous (F) GVHD with delayed anti-Dll1/4 administration. *P < .01 (1-way ANOVA) for TCD BM and group receiving early anti-Dll1/4 vs groups receiving isotype control or delayed anti-Dll1/Dll4 (n = 12, cumulative data from 3 experiments).
To evaluate whether delayed targeting of Dll1/4-mediated Notch signals impacted disease in the B10.D2→BALB/c model, we inhibited Dll1/Dll4 after day 40 (at the peak of Scl-cGVHD) or starting on day 10 after allo-HCT (before development of Scl-cGVHD signs). While no clear benefits were observed with either strategy of delayed blockade (Figure 2C), histopathological analysis revealed a trend toward decreased target organ injury and collagen deposition in animals treated with anti-Dll1/Dll4 from days 40 to 50 (P = .077; Figure 2D; supplemental Figure 4B-C). As our recent data showed a critical role for Notch blockade in the immediate peritransplant period in aGVHD,22 we tested the impact of briefly delaying our therapeutic intervention on Scl-cGVHD. As in aGVHD, a single peritransplant anti-Dll1/Dll4 injection was sufficient to induce long-term protection, which was mostly eliminated if the intervention was delayed by 48 hours (Figure 2E-F). These data identify the importance of Dll1/Dll4–mediated Notch signaling in the immediate posttransplant period for subsequent cGVHD pathogenesis.
Vβ3+ alloantigen-specific T cells are pathogenic in Scl-cGVHD
Since early Notch activity appeared essential for Scl-cGVHD pathogenesis, we studied pathogenic T cells during the immediate posttransplant window. Unlike other cGVHD models, the B10.D2→BALB/c model offered an opportunity to identify a population of alloantigen-specific T cells, since Vβ3+CD4+ T cells recognizing the Mtv6-encoded Mls3a minor alloantigen expressed in BALB/c, but not in donor B10.D2 tissues, can be tracked by flow cytometry (supplemental Figure 5A).45 Lethally irradiated BALB/c, but not syngeneic B10.D2, recipients of B10.D2-Thy1.1 splenocytes had activated Vβ3+ T cells (CD69 upregulation), consistent with an alloantigen-driven T-cell response (supplemental Figure 5B). In secondary lymphoid organs, Vβ3+ but not other tested CD4+ T cell Vβ families rapidly expanded starting day 2 posttransplant, peaked at day 6, and subsequently contracted (Figure 3A; supplemental Figure 5C), consistent with the pattern of a superantigen-driven response.45 Dll1/Dll4 blockade had no effect on posttransplant expansion of Vβ3+CD4+ alloreactive T cells, suggesting preservation of early alloantigen-driven T-cell proliferation (supplemental Figure 5C-D).
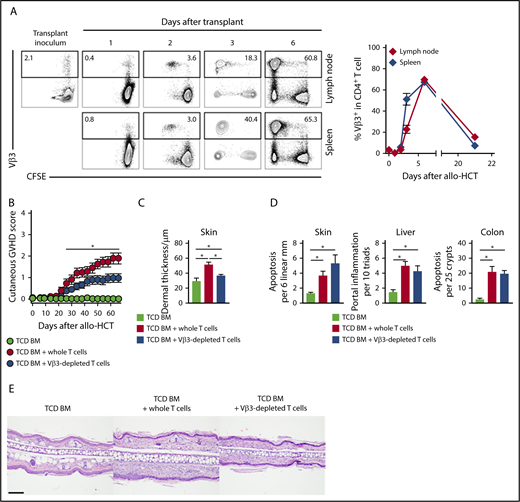
In the Scl-cGVHD model, immunodominant and pathogenic alloreactive Vβ3+T cells expand rapidly post-transplant and their elimination mitigates Scl-cGVHD. BALB/c mice were lethally irradiated and transplanted with B10.D2-Thy1.2 TCD BM (107 cells) supplemented with 12 × 106 CFSE-labeled B10.D2-Thy1.1 WT splenocytes. (A) Expression of Vβ3 in donor-derived CD90.1+CD4+ T cells in spleen or lymph nodes at the indicated time points after transplant. Vβ3+ T cells proliferated and expanded rapidly to become the majority of donor T cells on day 6, before their subsequent contraction. Representative flow cytometry plots, with numbers indicating the percentage of Vβ3+ cells among donor-derived CD4+ T cells (left). (B-C) B10.D2→BALB/c chimeras were generated by transplanting TCD BM only (light green circle) vs TCD BM with 3 × 106 WT T cells depleted of Vβ3+ T cells (blue circle) or sham manipulated (red circle). (B) Cutaneous cGVHD manifestations were monitored biweekly. Note significant cutaneous GVHD score reduction in mice receiving Vβ3-depleted T cells as compared with whole T-cell recipients. *P < .05 for all comparisons (1-way ANOVA). (C-D) Histopathologic analysis of animals depicted in panel (B). (C-D) Cumulative data show decreased pathological hallmarks of cGVHD (C) with transplantation of Vβ3-depleted T cells, without corresponding impact on the severity of aGVHD (D). *P < .05 (1-way ANOVA). (E) Representative microphotographs of hematoxylin and eosin–stained skin sections showing increased dermal thickness in the whole T cell as compared with TCD BM recipients, with decreased thickness in recipients of Vβ3-depleted T cells (ear lobes, day 60). Scale bar represents 100 µm.
In the Scl-cGVHD model, immunodominant and pathogenic alloreactive Vβ3+T cells expand rapidly post-transplant and their elimination mitigates Scl-cGVHD. BALB/c mice were lethally irradiated and transplanted with B10.D2-Thy1.2 TCD BM (107 cells) supplemented with 12 × 106 CFSE-labeled B10.D2-Thy1.1 WT splenocytes. (A) Expression of Vβ3 in donor-derived CD90.1+CD4+ T cells in spleen or lymph nodes at the indicated time points after transplant. Vβ3+ T cells proliferated and expanded rapidly to become the majority of donor T cells on day 6, before their subsequent contraction. Representative flow cytometry plots, with numbers indicating the percentage of Vβ3+ cells among donor-derived CD4+ T cells (left). (B-C) B10.D2→BALB/c chimeras were generated by transplanting TCD BM only (light green circle) vs TCD BM with 3 × 106 WT T cells depleted of Vβ3+ T cells (blue circle) or sham manipulated (red circle). (B) Cutaneous cGVHD manifestations were monitored biweekly. Note significant cutaneous GVHD score reduction in mice receiving Vβ3-depleted T cells as compared with whole T-cell recipients. *P < .05 for all comparisons (1-way ANOVA). (C-D) Histopathologic analysis of animals depicted in panel (B). (C-D) Cumulative data show decreased pathological hallmarks of cGVHD (C) with transplantation of Vβ3-depleted T cells, without corresponding impact on the severity of aGVHD (D). *P < .05 (1-way ANOVA). (E) Representative microphotographs of hematoxylin and eosin–stained skin sections showing increased dermal thickness in the whole T cell as compared with TCD BM recipients, with decreased thickness in recipients of Vβ3-depleted T cells (ear lobes, day 60). Scale bar represents 100 µm.
To determine whether Vβ3+ T cells were pathogenic in Scl-cGVHD, we depleted Vβ3+ T cells from the T-cell inoculum (>95% efficiency; data not shown). Transplantation of T cells depleted of Vβ3+ cells diminished the skin-specific Scl-cGVHD injury, without impacting the severity of systemic GVHD (Figure 3B and data not shown). This preclinical phenotype correlated with histopathological evaluation, as Vβ3-depleted recipients showed diminished dermal thickness but persisting features of aGVHD (Figure 3C-E). Flow cytometric analysis showed that in Vβ3-depleted recipients, the Vβ3neg T-cell fraction acquired high amounts of proinflammatory cytokines (supplemental Figure 5E). Thus, alloantigen-specific Vβ3+ T cells contributed to Scl-cGVHD pathogenesis, although other T cells could compensate for their loss to induce GVHD.
Notch controls early posttransplant effector functions in alloantigen-specific Vβ3+ T cells
The Scl-cGVHD model provided a unique opportunity to study the effects of Notch signaling in a defined alloantigen-specific T-cell population. Control or Notch-deprived T cells were retrieved from B10.D2→BALB/c chimeras early after allo-HCT for immunophenotypic and transcriptional analysis. We focused on day 2, given our results of dominant Notch activity during that time, and on day 6, a time point when the immunophenotypic effects of Notch blockade are recognizable and predictive of preclinical effectiveness.
Alloreactive T cells showed similar activation at early stages, independent of Notch signaling, as seen by the surface expression of CD25, CD43, and CD69 on day 2 after allo-HCT (Figure 4A and data not shown). On day 6, Notch blockade led to decreased CD25 expression. However, both WT and pan-Notch–inhibited Vβ3+CD4+ and CD8+ T cells showed similar in vivo proliferation as assessed with eFluor450 dilution (Figure 4B).
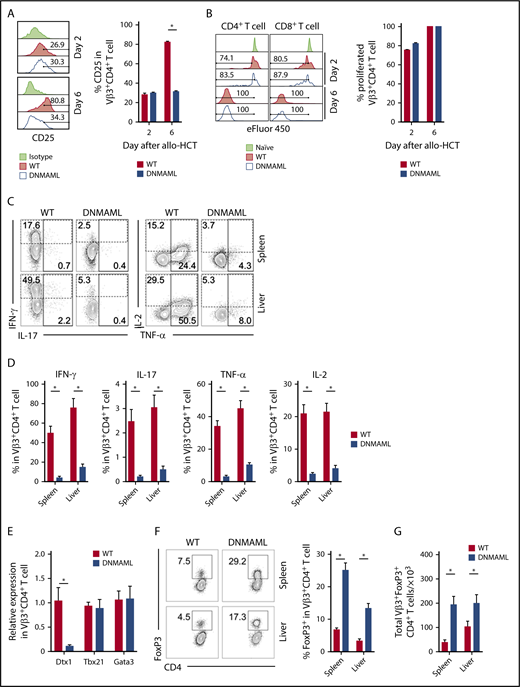
Notch signaling enhances proinflammatory functions of alloreactive T cells and suppresses the expansion of alloantigen-specific regulatory T cells after transplantation. BALB/c recipients were lethally irradiated (800 cGy) and transplanted with B10.D2-Thy1.2 TCD BM (107 cells) plus 12 × 106 B10.D2-Thy1.1 WT or B10.D2-DNMAML splenocytes (labeled with eFluor450 to allow for tracking of cell division). Tissues were retrieved for flow cytometric analysis of alloantigen-specific Vβ3+ T-cell populations. (A) CD25 expression in CD4+Vβ3+ T cells at days 2 and 6 after transplantation. *P < .01 (2-tailed unpaired Student t test). (B) eFluor450 dilution at days 2 and 6 posttransplant showing preserved early proliferation of DNMAML as compared with WT CD4+Vβ3+ T cells. (C-D) Flow cytometric analysis of cytokine production by WT vs Notch-deprived DNMAML CD4+Vβ3+ T cells isolated from spleen or liver at day 6 after allo-HCT (ex vivo anti-CD3/CD28 restimulation before staining for intracellular cytokines). Representative contour plots are shown in panel C and cumulative data in panel D. Numbers indicate the percentage of events falling within indicated rectangular gates. *P < .01 (2-tailed unpaired Student t test). (E) Notch inhibition in Vβ3+CD4+ T cells alters transcription of Notch targets but does not affect Th-lineage polarization. RNA was isolated; a complementary DNA library was generated from sort-purified Vβ3+CD4+ T cells on day 6 after allo-HCT and then subjected to quantitative reverse-transcription PCR. While Notch targets were depressed (Dtx1), canonical Th1 (Tbx21) and Th2 (Gata3) transcription factors remained unaffected. (F-G) Notch inhibition enhances expansion of Vβ3+CD4+FoxP3+ Tregs after allo-HCT. Data show expression of FoxP3 in Vβ3+CD4+ T cells 6 days after allo-HCT (F) and total Vβ3+ Tregs at that time point (G). *P < .01 (2-tailed unpaired Student t test). Data in panels A-F are representative of at least 2 experiments with 10 animals per cohort.
Notch signaling enhances proinflammatory functions of alloreactive T cells and suppresses the expansion of alloantigen-specific regulatory T cells after transplantation. BALB/c recipients were lethally irradiated (800 cGy) and transplanted with B10.D2-Thy1.2 TCD BM (107 cells) plus 12 × 106 B10.D2-Thy1.1 WT or B10.D2-DNMAML splenocytes (labeled with eFluor450 to allow for tracking of cell division). Tissues were retrieved for flow cytometric analysis of alloantigen-specific Vβ3+ T-cell populations. (A) CD25 expression in CD4+Vβ3+ T cells at days 2 and 6 after transplantation. *P < .01 (2-tailed unpaired Student t test). (B) eFluor450 dilution at days 2 and 6 posttransplant showing preserved early proliferation of DNMAML as compared with WT CD4+Vβ3+ T cells. (C-D) Flow cytometric analysis of cytokine production by WT vs Notch-deprived DNMAML CD4+Vβ3+ T cells isolated from spleen or liver at day 6 after allo-HCT (ex vivo anti-CD3/CD28 restimulation before staining for intracellular cytokines). Representative contour plots are shown in panel C and cumulative data in panel D. Numbers indicate the percentage of events falling within indicated rectangular gates. *P < .01 (2-tailed unpaired Student t test). (E) Notch inhibition in Vβ3+CD4+ T cells alters transcription of Notch targets but does not affect Th-lineage polarization. RNA was isolated; a complementary DNA library was generated from sort-purified Vβ3+CD4+ T cells on day 6 after allo-HCT and then subjected to quantitative reverse-transcription PCR. While Notch targets were depressed (Dtx1), canonical Th1 (Tbx21) and Th2 (Gata3) transcription factors remained unaffected. (F-G) Notch inhibition enhances expansion of Vβ3+CD4+FoxP3+ Tregs after allo-HCT. Data show expression of FoxP3 in Vβ3+CD4+ T cells 6 days after allo-HCT (F) and total Vβ3+ Tregs at that time point (G). *P < .01 (2-tailed unpaired Student t test). Data in panels A-F are representative of at least 2 experiments with 10 animals per cohort.
At day 6 after transplant, Notch blockade suppressed proinflammatory cytokine production (Figure 4C-D), as seen in polyclonal T cells inducing aGVHD.19-22 Notch blockade did not influence expression of genes encoding the transcription factors T-bet and Gata3, while expression of Dtx1, a known Notch target gene, was decreased (Figure 4E). T-bet protein levels were not affected upon disruption of the Notch transcriptional complex by DNMAML, although Dll1/Dll4 blockade decreased T-bet abundance (supplemental Figure 6A-B). These findings suggest that noncanonical or non–cell-autonomous effects of Notch contribute to T-bet expression, although the dominant pathogenic effects of Notch were mediated by canonical MAML-dependent mechanisms. Concurrently, Notch ablation led to increased FoxP3+ Treg accumulation within the Vβ3+CD4+ T cells (Figure 4F-G). Thus, in the Scl-cGVHD model, Notch blockade regulated selected effector functions of alloreactive effector T cells and favored expansion of alloantigen-specific Tregs.
Notch blockade in T cells prevents BO-cGVHD development
We next sought to test the effects of Notch signaling in a complementary MHC-mismatched model of multi–organ system cGVHD with dominant BO. In this model, a low dose of C57BL/6 T cells is transplanted into B10.BR recipients, inducing limited inflammation and aGVHD but subsequent tissue damage and complex immune dysregulation, including a follicular T helper cell (Tfh) response and a germinal center (GC) B-cell reaction. Ultimately, BO-cGVHD leads to fibrosis in a subset of the terminal bronchioles and quantifiable obstructive airway disease.27
To evaluate the role of Notch signaling in BO-cGVHD, we first transplanted B10.BR mice with B6 WT TCD BM vs TCD BM with 80 × 103 T cells from control B6 (WT) or B6-DNMAML donors. Animals were monitored and subjected to pulmonary function analysis on day 56. DNMAML T-cell transfer effectively prevented clinical BO-cGVHD manifestations as shown by pulmonary resistance, elastance, and compliance that were indistinguishable from those in TCD BM recipients (Figure 5A). Thus, Notch signaling in T cells was critical to induce key clinical BO-cGVHD manifestations.
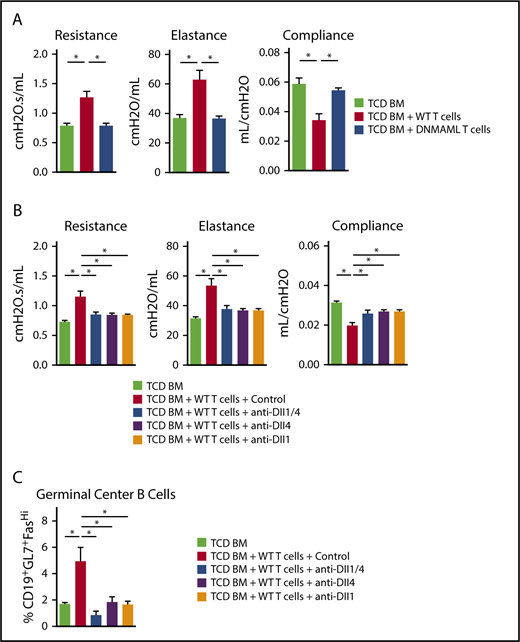
Notch signaling mediated by Dll1 and Dll4 is necessary for the development and persistence of BO-cGVHD. B10.BR recipients were conditioned with total body irradiation (8.3 Gy, day −1) and cyclophosphamide (120 mg/kg/day i.p., days −3 and −2) before transplantation with B6 WT TCD BM only (107 cells) vs TCD BM plus 80 × 103 B6 WT or B6-DNMAML T cells. (A) Pulmonary function testing (resistance, elastance, and compliance) was performed at day 56 after allo-HCT. Recipients of WT, but not DNMAML, T cells showed profound changes consistent with BO-cGVHD airway disease. *P < .05 (1-way ANOVA). (B) B10.BR recipients were transplanted as indicated above. After 4 weeks, systemic treatment was started with isotype control vs anti-Dll1, anti-Dll4, or a combination of anti-Dll1 and anti-Dll4 antibodies (5 mg/kg i.p. twice weekly, days 28-56). Pulmonary function tests were performed at day 56. *P < .05 for mice treated with isotype control antibodies (BO-cGVHD) vs mice receiving only TCD BM (GVHD-free control) and all groups receiving Notch ligand neutralizing antibodies. (C) Dll1 and/or Dll4 inhibition alters GC B-cell homeostasis, significantly decreasing the frequency of B cells with a GL7+Fashi GC B-cell phenotype in the spleen. *P < .05. Representative data from one of 2 experiments is shown with 8 animals per group.
Notch signaling mediated by Dll1 and Dll4 is necessary for the development and persistence of BO-cGVHD. B10.BR recipients were conditioned with total body irradiation (8.3 Gy, day −1) and cyclophosphamide (120 mg/kg/day i.p., days −3 and −2) before transplantation with B6 WT TCD BM only (107 cells) vs TCD BM plus 80 × 103 B6 WT or B6-DNMAML T cells. (A) Pulmonary function testing (resistance, elastance, and compliance) was performed at day 56 after allo-HCT. Recipients of WT, but not DNMAML, T cells showed profound changes consistent with BO-cGVHD airway disease. *P < .05 (1-way ANOVA). (B) B10.BR recipients were transplanted as indicated above. After 4 weeks, systemic treatment was started with isotype control vs anti-Dll1, anti-Dll4, or a combination of anti-Dll1 and anti-Dll4 antibodies (5 mg/kg i.p. twice weekly, days 28-56). Pulmonary function tests were performed at day 56. *P < .05 for mice treated with isotype control antibodies (BO-cGVHD) vs mice receiving only TCD BM (GVHD-free control) and all groups receiving Notch ligand neutralizing antibodies. (C) Dll1 and/or Dll4 inhibition alters GC B-cell homeostasis, significantly decreasing the frequency of B cells with a GL7+Fashi GC B-cell phenotype in the spleen. *P < .05. Representative data from one of 2 experiments is shown with 8 animals per group.
We next used Dll1/Dll4 neutralization to identify individual Notch ligands involved in BO-cGVHD and potential therapeutic targets. Allo-HCT recipients were observed for 4 weeks to allow for BO-cGVHD development before starting 4 biweekly injections with anti-Dll1, anti-Dll4, both anti-Dll1 and anti-Dll4, or a control mAb. While WT T-cell recipients treated with control mAb showed impaired pulmonary function consistent with BO, anti-Dll1/Dll4–treated mice demonstrated almost complete reversal of the clinical phenotype (Figure 5B). Targeting individual Notch ligands Dll1 or Dll4 was sufficient to offset BO in WT recipients (Figure 5B), indicating nonredundant roles of Dll1 and Dll4 in BO-cGVHD. In this model, Notch inhibition via Dll1- or Dll4-neutralizing mAbs also decreased the accumulation of B cells expressing GL7, suggestive of GC origin (Figure 5C).
Altogether, Notch signaling remained pathogenic throughout the disease process in BO-cGVHD. Individual Delta-like ligands were key mediators of signaling, with nonredundant roles of individual ligands, and could be safely targeted to mitigate even established cGVHD.
Notch2 receptor is a therapeutic target in BO-cGVHD
Notch1 and Notch2 in T cells are required to mediate aGVHD and Scl-cGVHD, with a dominant role for Notch1. To identify the key Notch receptors involved in BO-cGVHD, we first relied on C57BL/6 mice lacking individual Notch receptors in T cells.21 B10.BR conditioned mice received WT, Notch1-deficient, Notch2-deficient, or Notch1/2-deficient T cells. At day 49, pulmonary function tests showed marked improvement in recipients of T cells lacking Notch1, Notch2, or both receptors (Figure 6A). GC B-cell populations were reduced with either Notch1 and/or Notch2 loss (Figure 6B). All strategies decreased the relative abundance of Tfh cells while enhancing accumulation of T follicular regulatory (Tfr) cells, as shown through an increased Tfr/Tfh ratio in recipients of Notch-deficient T cells (Figure 6C).
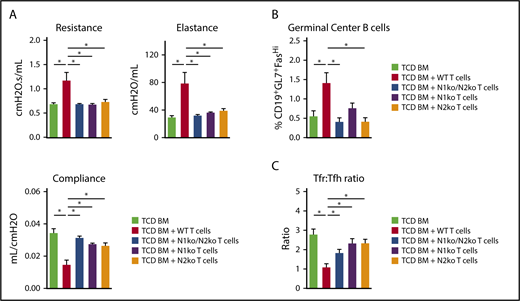
Notch1 and Notch 2 receptors deliver nonredundant pathogenic signals in T cells during BO-cGVHD. B10.BR recipients were conditioned with irradiation and cyclophosphamide and transplanted with B6 WT TCD BM cells (107) supplemented with 80 × 103 B6 WT, Cd4-Cre × Notch1f/f (N1Ko), Cd4-Cre × Notch2f/f (N2Ko), and Cd4-Cre×Notch1f/fNotch2f/f (N1Ko/N2Ko) T cells. (A) Notch1 and Notch2 receptors are nonredundant in driving pathogenic Notch signaling in T cells to cause BO-cGVHD. Pulmonary function tests at day 49 showing preservation of pulmonary functions in mice receiving Notch1-deficient, Notch2-deficient, or Notch1/2-deficient vs WT T cells. *P < .05 (1-way ANOVA). (B-C) Notch1 and/or Notch2 loss in T cells decreases the accumulation of GC B-cell (B) and Tfh cell (C) posttransplant in spleens of recipient animals. Spleens were harvested on day 60 posttransplant and analyzed for GC B cells as well as Tfh and Tfr cells. The Tfr/Tfh ratio was calculated by dividing the percentages of respective subsets in parental populations. *P < .05 (1-way ANOVA).
Notch1 and Notch 2 receptors deliver nonredundant pathogenic signals in T cells during BO-cGVHD. B10.BR recipients were conditioned with irradiation and cyclophosphamide and transplanted with B6 WT TCD BM cells (107) supplemented with 80 × 103 B6 WT, Cd4-Cre × Notch1f/f (N1Ko), Cd4-Cre × Notch2f/f (N2Ko), and Cd4-Cre×Notch1f/fNotch2f/f (N1Ko/N2Ko) T cells. (A) Notch1 and Notch2 receptors are nonredundant in driving pathogenic Notch signaling in T cells to cause BO-cGVHD. Pulmonary function tests at day 49 showing preservation of pulmonary functions in mice receiving Notch1-deficient, Notch2-deficient, or Notch1/2-deficient vs WT T cells. *P < .05 (1-way ANOVA). (B-C) Notch1 and/or Notch2 loss in T cells decreases the accumulation of GC B-cell (B) and Tfh cell (C) posttransplant in spleens of recipient animals. Spleens were harvested on day 60 posttransplant and analyzed for GC B cells as well as Tfh and Tfr cells. The Tfr/Tfh ratio was calculated by dividing the percentages of respective subsets in parental populations. *P < .05 (1-way ANOVA).
Thus, both Notch1 and Notch2 receptors could be considered therapeutic targets. However, Notch1 plays an essential role in intestinal stem cell homeostasis, and its targeting is associated with systemic toxicity, including after allo-HCT.21,46 Therefore, we focused on achieving Notch blockade with anti-NRR2 antibodies in BO-cGVHD. Notch2 blockade prevented the BO-cGVHD phenotype, as seen through intact pulmonary function test results (Figure 7A) and decreased bronchiolar collagen deposition, a hallmark of BO-cGVHD (Figure 7B-C). Furthermore, anti-NRR2–treated animals showed decreased average size of splenic germinal centers and increased follicular regulatory T cells (Figure 7D-E and data not shown). Prolonged anti-NRR2 therapy was well tolerated and allowed steady weight recovery after allo-HCT (supplemental Figure 7).
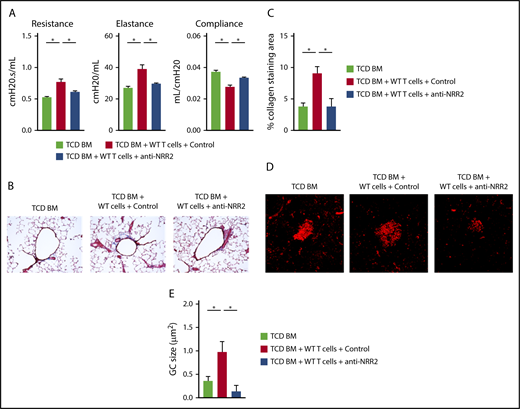
Notch2 inhibition is a potential therapeutic strategy in BO-cGVHD without the detrimental side effects characteristic of Notch1 targeting. Following conditioning, B10.BR recipients were transplanted with B6 WT TCD BM cells (107 cells) supplemented with 74 × 103 B6 WT T cells. Treatment with control vs anti-NRR2 antibodies was given with biweekly injections for 7 weeks, before pulmonary testing was completed. (A) Pulmonary functions tests at day 49 after allo-HCT showing beneficial effects of Notch2 blockade with anti-NRR2 antibodies. (B-C) Anti-NRR2 blockade decreases peribronchiolar collagen deposition in BO-cGVHD. Representative microphotographs of Masson’s trichrome stained lung sections taken at ×200 magnification (B). Cumulative data (C) showing quantification of collagen deposition expressed as a ratio of blue over total stained area. (D-E) Peanut agglutinin staining in the spleens of TCD BM recipients vs recipients of TCD BM plus T cells treated with isotype control or anti-NRR2 antibodies show dampened GC B-cell responses in the spleens of anti-NRR2 treated recipients. Representative microphotographs taken at ×200 magnification (D) and cumulative data (E) quantifying average GC size. *P < .05 (1-way ANOVA).
Notch2 inhibition is a potential therapeutic strategy in BO-cGVHD without the detrimental side effects characteristic of Notch1 targeting. Following conditioning, B10.BR recipients were transplanted with B6 WT TCD BM cells (107 cells) supplemented with 74 × 103 B6 WT T cells. Treatment with control vs anti-NRR2 antibodies was given with biweekly injections for 7 weeks, before pulmonary testing was completed. (A) Pulmonary functions tests at day 49 after allo-HCT showing beneficial effects of Notch2 blockade with anti-NRR2 antibodies. (B-C) Anti-NRR2 blockade decreases peribronchiolar collagen deposition in BO-cGVHD. Representative microphotographs of Masson’s trichrome stained lung sections taken at ×200 magnification (B). Cumulative data (C) showing quantification of collagen deposition expressed as a ratio of blue over total stained area. (D-E) Peanut agglutinin staining in the spleens of TCD BM recipients vs recipients of TCD BM plus T cells treated with isotype control or anti-NRR2 antibodies show dampened GC B-cell responses in the spleens of anti-NRR2 treated recipients. Representative microphotographs taken at ×200 magnification (D) and cumulative data (E) quantifying average GC size. *P < .05 (1-way ANOVA).
Taken together, Notch signaling driving BO-cGVHD was mediated nonredundantly by Notch1/Notch2 receptors and Dll1/Dll4 ligands. Pathogenic Notch signals persisted over time in BO-cGVHD, and immunologic mediators of disease remained exquisitely sensitive to systemic targeting of Dll1, Dll4, or Notch2 in this preclinical model of cGVHD.
Discussion
cGVHD remains prevalent after transplant, inflicting significant long-term morbidity and mortality on patients.47 We previously discovered a critical pathogenic role for Notch signaling in T cells during aGVHD.19-22 Here, we employ established preclinical cGVHD models mimicking human disease phenotypes: a model with systemic fibrotic injury arising after clinically apparent aGVHD (Scl-cGVHD), and a less inflammatory model leading to systemic cGVHD with prominent lung involvement (BO-cGVHD). Our strategy was based on past experience with preclinical cGVHD studies that highlight the importance of studying complementary models to better evaluate distinct aspects of this complex disease.9,28,29,48 Our work revealed the importance of Notch signaling at several stages of cGVHD pathogenesis.
In the Scl-cGVHD model, Notch signaling was driven predominantly by a single Notch ligand (Dll4). By targeting Dll4 within the first 2 days after transplant, we achieved near-complete disease prevention. A 2-day delay in Notch ligand blockade, or intervention during the course of established disease, failed to induce clinical protection, although a trend toward decreased tissue injury was observed. This narrow window of Dll4-driven Notch activity in Scl-cGVHD is reminiscent of our prior observations in MHC-mismatched aGVHD models. Mechanistically, our findings might be explained by the characteristics and kinetics of the posttransplant alloresponse in the Scl-cGVHD model. In Scl-cGVHD, early robust alloreactive Vβ3+ T-cell expansion shares features with T-cell responses seen in aGVHD while leading subsequently to cGVHD.49
To document specific effects of Notch blockade in pathogenic effector T cells contributing to Scl-cGVHD, we tracked alloreactive Vβ3+ T cells. While Notch-deprived Vβ3+ alloreactive T cells became activated, proliferated, and expanded in vivo, Notch blockade induced major immunomodulatory effects, decreasing the production of proinflammatory cytokines and enhancing Treg expansion. The increased sensitivity of Tregs to IL-2 (a feature capitalized upon with low-dose IL-2 in human cGVHD50 ) might be important to sustain Treg expansion despite a low IL-2 environment in the setting of Notch inhibition (Figures 1F and 4F-G; supplemental Figure 2E). Beyond immune regulation, Notch might also play a role in fibrogenesis in cGVHD, as its role in this process has been observed in different diseases.51 However, existing evidence would favor a role for Jagged, but not Delta-like ligands, as putative mediators of Notch activity in fibrosis.52,53
Our observations in BO-cGVHD complement our findings in Scl-cGVHD. In both models, Notch signaling was important for disease manifestations, with differences in temporal requirements for Notch signals (early Notch1 and Dll4 in Scl-cGVHD and longitudinal and nonredundant Notch1, Notch2, Dll1, and Dll4 in BO-cGVHD). As in other GVHD models tested so far, peritransplant Notch signaling in T cells was critical for induction of the BO-cGVHD phenotype, with Notch ablation regulating Tfh and GC B-cell homeostasis, both of which are essential in cGVHD immunopathology. Other aspects of B-cell immunity independent of GCs could also be at play.49 Fibroblastic stromal cells, which are essential to drive Notch signaling in aGVHD,22 are also sources of B-cell–activating factor, a trophic and pathogenic factor in cGVHD.54 Whether these cells also provide Notch inputs during cGVHD remains to be seen. In aGVHD, the Dll4-Notch1 axis was responsible for the majority of Notch effects in T cells. In BO-cGVHD, we discovered nonredundant roles for Notch1 and Notch2 receptors, as well as Dll1 and Dll4 ligands. This phenomenon suggests that nonoverlapping cellular sources of Notch ligands could be important or that T cells are sensitive only to partial loss of Notch signaling in BO-cGVHD. This observation broadens the range of Notch pathway members that could be considered for therapeutic blockade.
Our findings add to a growing body of work identifying the early posttransplant environment as critical for subsequent cGVHD.11-15 The consequences of Notch blockade early after allo-HCT bear similarities with posttransplant cyclophosphamide, with partly overlapping mechanisms,55 yet broader effects of Notch blockade on T-cell homeostasis and dysregulated humoral alloresponse. Moreover, our observations also identify pathogenic Notch activity contributing to cGVHD injury long after resolution of early posttransplant inflammation. Our work does not tease apart the Notch-dependent cellular subsets responsible for ongoing cGVHD, but together with the recent discovery of Notch2-BCR axis hyperactivity in human cGVHD,24 it points to the importance of dysregulated B-cell responses.
Our studies add to the increasing level of evidence supporting a central role for canonical Notch signaling in GVHD. Identifying Notch targeting as an effective strategy to prevent and/or treat cGVHD in mouse models raises hope to mitigate the most difficult forms of cGVHD seen in humans.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases (grant R01-AI091627) (I.M.) (grants R01-AI34495 and P01 AI056299) (B.R.B.); National Institutes of Health, National Cancer Institute (grant P01-CA142106) (B.R.B.); National Institutes of Health, National Heart, Lung, and Blood Institute (grant R01-HL129061) (S.S.); and the Leukemia and Lymphoma Society (grant CDP 1227-14) (I.M.) (grant TRP 6462-15) (I.M. and B.R.B.) (grant TRP 6497-16) (S.S.). Individual support included a research training award from the American Society of Hematology (V.R.), a Career Development Award from the American Society for Blood and Marrow Transplantation (V.R.), Huntsman Cancer Foundation funding (V.R.), the National Institute of General Medical Sciences (grant T32-GM007315) (J.C.) (grant T32-GM007863) (J.C. and E.P.).
Authorship
Contribution: V.R. designed and performed experiments, analyzed data and wrote the manuscript; K.P., J.C., J.D., E.T.P., R.F., S.I., M.Z., and A.F. performed experiments and analyzed data; M.A.P. performed blinded histopathological analysis; S.S. provided collaborative advice regarding B-cell responses in cGVHD; M.Y. and C.W.S. contributed key reagents; B.R.B. and I.M. designed experiments, supervised the study, and wrote the manuscript; and all authors reviewed the final version of the paper.
Conflict-of-interest disclosure: M.Y. and C.W.S. are employees of Genentech, Inc. The remaining authors declare no competing financial interests.
Correspondence: Bruce R. Blazar, MMC 109, 420 Delaware St SE, University of Minnesota, Minneapolis, MN 55455; e-mail: blaza001@umn.edu; and Ivan Maillard, 451 BRB2/3, 421 Curie Blvd, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104; e-mail: imaillar@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal