Key Points
Eltrombopag/azacitidine was inferior to placebo/azacitidine in higher-risk MDS patients with respect to platelet-related and survival end points.
Findings from this study do not indicate a role for combining eltrombopag with azacitidine in patients with intermediate/high-risk MDS.

Abstract
Azacitidine treatment of myelodysplastic syndromes (MDSs) generally exacerbates thrombocytopenia during the first treatment cycles. A Study of Eltrombopag in Myelodysplastic Syndromes Receiving Azacitidine (SUPPORT), a phase 3, randomized, double-blind, placebo-controlled study, investigated the platelet supportive effects of eltrombopag given concomitantly with azacitidine. International Prognostic Scoring System intermediate-1, intermediate-2, or high-risk MDS patients with baseline platelets <75 × 109/L were randomized 1:1 to eltrombopag (start, 200 mg/d [East Asians, 100 mg/d], maximum, 300 mg/d [East Asians, 150 mg/d]) or placebo, plus azacitidine (75 mg/m2 subcutaneously once daily for 7 days every 28 days). The primary end point was the proportion of patients platelet transfusion-free during cycles 1 through 4 of azacitidine therapy. Based on planned interim analyses, an independent data monitoring committee recommended stopping the study prematurely because efficacy outcomes crossed the predefined futility threshold and for safety reasons. At termination, 28/179 (16%) eltrombopag and 55/177 (31%) placebo patients met the primary end point. Overall response (International Working Group criteria; complete, marrow, or partial response) occurred in 20% and 35% of eltrombopag and placebo patients, respectively, by investigator assessment. There was no difference in hematologic improvement in any cell lineage between the 2 arms. There was no improvement in overall or progression-free survival. Adverse events with ≥10% occurrence in the eltrombopag vs placebo arm were febrile neutropenia and diarrhea. Compared with azacitidine alone, eltrombopag plus azacitidine worsened platelet recovery, with lower response rates and a trend toward increased progression to acute myeloid leukemia. This trial was registered at www.clinicaltrials.gov as #NCT02158936.
Introduction
Myelodysplastic syndromes (MDSs) are clonal malignancies of pluripotent hematopoietic stem cells, characterized by ineffective hematopoiesis leading to cytopenias.1 Thrombocytopenia occurs in 40% to 65% of patients with MDS and predicts worse prognosis.2,3 Hypomethylating agents, predominantly azacitidine, represent the current standard of care as the first-line treatment of patients with MDS and a higher risk of transformation to acute myeloid leukemia (AML).4,5 However, hypomethylating agents are generally associated with the development or exacerbation of thrombocytopenia, particularly in the first few cycles, before disease response when thrombocytopenia is expected to improve.6,7 Thrombocytopenia is burdensome to the patient because of requirements for more intensive monitoring during treatment and the higher risk of bleeding. Azacitidine dose adjustments may compromise efficacy outcomes; therefore, treatments that can alleviate hematologic toxicity and improve platelet counts in this setting may ameliorate patient morbidity.
Eltrombopag is an oral, nonpeptide, thrombopoietin receptor agonist (TPO-RA) that stimulates platelet production and is approved for the treatment of patients with chronic immune thrombocytopenia, hepatitis C virus-related thrombocytopenia, and refractory severe aplastic anemia.8 Eltrombopag binds to the transmembrane domain of the TPO-RA, inducing proliferation and differentiation of bone marrow (BM) progenitor cells in the megakaryocyte lineage.9,10
Improved platelet counts following treatment with eltrombopag have been observed in trials of patients with immune thrombocytopenia and severe aplastic anemia.11-14 More recent clinical trials with single-agent eltrombopag at doses of ≤300 mg/day have also demonstrated improvements in platelet counts and reductions in the incidence of thrombocytopenic events in low-risk MDS, high-risk MDS, and AML.15-17 Results of phase 1 and 2 trials have suggested that the combination of eltrombopag with azacitidine is feasible and well tolerated.18,19 Additionally, a phase 1/2 study in lower risk MDS patients showed that the combination of another TPO-RA, romiplostim, with azacitidine was well tolerated, although blast cell counts were transiently increased in some patients.20 Here, we report the Study of Eltrombopag in Myelodysplastic Syndromes Receiving Azacitidine (SUPPORT), a phase 3 trial investigating the efficacy and safety of eltrombopag as platelet supportive care, in patients with intermediate- to high-risk MDS and thrombocytopenia who were receiving azacitidine.
Methods
Key inclusion/exclusion criteria
Eligible patients were aged ≥18 years (≥20 years in Taiwan) with a diagnosis of MDS using World Health Organization21 or French-American-British22 classifications, International Prognostic Scoring System (IPSS)23 intermediate-1 (Int-1), intermediate-2 (Int-2), or high-risk MDS (confirmed by investigator examination of bone marrow), and at least 1 platelet count <75 × 109/L within 28 days before the first azacitidine dose. Patients were also required to have an Eastern Cooperative Oncology Group status of 0 to 2 and adequate renal and hepatic function. Key exclusion criteria were previous treatment of MDS with hypomethylating agents or induction chemotherapy, and previous treatment with any TPO-RA.
Study design
This was a randomized, double-blind, placebo-controlled, international, multicenter, phase 3 study. Patients were centrally randomized 1:1 to eltrombopag or placebo, in combination with azacitidine. The randomization schedule was based on an in-house system generated by the GlaxoSmithKline Biostatistics Department and stratified by IPSS risk score (Int-1 vs Int-2 vs high-risk MDS), baseline platelet count (<50 × 109/L vs ≥50 × 109/L), and platelet transfusion dependence (defined as receiving at least 2 platelet transfusions in the 4 weeks before randomization; yes/no). Patients were treated for ≥6 cycles of azacitidine and continued treatment as long as benefit was derived or until disease progression, unacceptable toxicity, or death.
Azacitidine was administered subcutaneously at a dose of 75 mg/m2 once daily for 7 consecutive days, every 28 days, with treatment breaks of up to 3 days allowed (dose adjustments were permitted based on the country-specific product label). Patients received eltrombopag or placebo continuously from day 1 of azacitidine at a starting dose of 200 mg/d (100 mg/d for East Asians), adjusted by 100-mg increments (50-mg increments for East Asians) to a maximum of 300 mg/d (150 mg/d for East Asians) in an attempt to ensure that platelet counts remained sufficient to avoid platelet transfusions and bleeding events. Dose adjustments were based on platelet counts as follows: for platelet counts <100 × 109/L, doses were increased to the next increment on day 1 of a treatment cycle; for platelet counts 100 to 200 × 109/L, doses were maintained without change; for platelet counts >200 to 400 × 109/L, doses were decreased to the next lower dose level at any time (but no more frequently than every 2 weeks); and for platelet counts >400 × 109/L, doses were interrupted and reinitiated at the next lowest dose level when platelet counts reached <200 × 109/L. Any change in the dose of eltrombopag/placebo was followed by weekly hematology assessments for at least 2 consecutive weeks. The investigator or treating physician could remove the treatment assignment blinding only in the case of an emergency or in the event of a serious medical condition.
The study was conducted in accordance with the Declaration of Helsinki, and an independent ethics committee or institutional review board for each study site approved the study protocol. All patients provided written informed consent to participate in the trial.
Outcomes
The primary end point was the proportion of patients who were free of platelet transfusions during cycles 1 through 4 of azacitidine therapy. The study authors reasoned that the clinical benefit of eltrombopag would be most meaningful during the first 4 treatment cycles; this is when patients typically experience the highest thrombocytopenic risk and greatest platelet transfusion requirements before response to azacitidine, which is anticipated at a median of 4 months. Secondary end points included overall survival (OS), disease response (modified 2006 International Working Group [IWG]), duration of response, progression to AML and progression-free survival (PFS), hematologic improvement, safety, and tolerability.
Data were collected on platelet and red blood cell transfusions for 8 weeks before day 1, then from day 1 through to the 4-week, poststudy follow-up visit. Survival was assessed every 3 months after the 4-week, poststudy follow-up visit. Patients were to be followed for survival until at least completion of the primary analysis or until at least 275 events (deaths) occurred. Early termination of the study limited the execution of the planned survival follow-up (see “Results”) and it was not possible to follow patients until at least 275 events occurred. Disease progression and progression to AML were defined by the modified 2006 IWG criteria24 and assessed by the investigator on the first day of each cycle and at the end of therapy visit. BM biopsies were performed to confirm disease response and disease progression, and regularly every 6 months until disease progression or study withdrawal. Hematology assessments were conducted at least weekly during the first cycle, and on days 1 and 15 of cycles 2 through 6.
For this trial, progression to AML in patients with baseline BM blast <20% was defined as meeting the definition of disease progression according to the modified 2006 IWG response criteria for MDS, which requires ≥50% increase in blasts,24 with the additional requirement that BM blast or peripheral blast increased from <20% at baseline to ≥20% postbaseline. Patients with baseline BM blasts of 20% to 30% by central review were also classified as experiencing progression to AML if the definition of disease progression was met (according to the modified 2006 IWG response criteria for MDS, which require ≥50% increase in blasts), with the additional requirement that BM blasts or peripheral blast increase to >30% postbaseline. Central review of all BM examinations and peripheral blood smears were performed but were not used to inform investigator assessments or treatment decisions. Hematologic improvement for platelets, neutrophils, and hemoglobin was calculated based on the modified IWG criteria for MDS, to be observed for 56 days.
Adverse events (AEs) were assessed from the first dose of study treatment until the 4-week poststudy assessment, graded according to the National Cancer Institute Common Terminology Criteria for AEs (CTCAE), version 4.0, and coded using the Medical Dictionary for Regulatory Activities, version 19.0.
Next-generation sequencing methods
Next-generation sequencing of whole blood samples was conducted using a panel targeting 365 genes. Analyses were focused on a targeted subset of 53 genes frequently mutated in MDS and a further subset of 18 genes associated with poor prognosis.25-30 The relationship between the 2 arms and the number of carriers vs noncarriers in any of 53 genes frequently mutated in MDS (and 18 genes associated with poor prognosis) at baseline were tested by Fisher exact test.
Statistical analyses
A total of 350 patients (175 for each treatment arm) were required to provide 90% power to demonstrate a statistically significant difference in the proportion of patients who were platelet transfusion–independent during cycles 1 through 4 between eltrombopag and placebo (assumed to be 50% and 30%, respectively),20,31 with a 2-sided 5% level of significance. For the OS end point, 275 events were required to provide 80% power.
The proportion of patients who were transfusion independent were compared between treatment arms using a stratified Cochran-Mantel-Haenszel χ2 test adjusting for stratification variables, with 95% confidence interval (95% CI) for the odds ratio (OR). The key secondary end point of OS was tested using a hierarchical procedure, based on first showing a significant benefit in favor of eltrombopag for the primary end point. OS was summarized by treatment groups via Kaplan-Meier curves with 95% CI, and treatment arms were compared using a stratified log-rank test accounting for stratification variables. PFS, defined as the time from randomization until either disease progression or death, was summarized by treatment groups through Kaplan-Meier curves with 95% CI, and treatment arms were compared using a stratified log-rank test accounting for stratification variables. The proportion of patients with progression to AML, overall response (complete response [CR], marrow CR, or partial response), and with hematologic improvement (platelets, neutrophils, and hemoglobin) were summarized by treatment and compared between treatment groups using Cochran-Mantel-Haenszel tests accounting for stratification variables, with 95% CI for ORs.
Two interim analyses were planned. One was for safety, which occurred after 70 patients (20% of the target sample) had completed 4 cycles of treatment, withdrawn, or died. The second, for safety and futility of the primary end point of transfusion independence, was conducted after 140 patients (40% of the target sample) had completed 4 cycles of treatment, withdrawn, or died. Futility was to be determined according to the primary end point, with a 1-sided threshold of P > .9.
Results
Study population and disposition
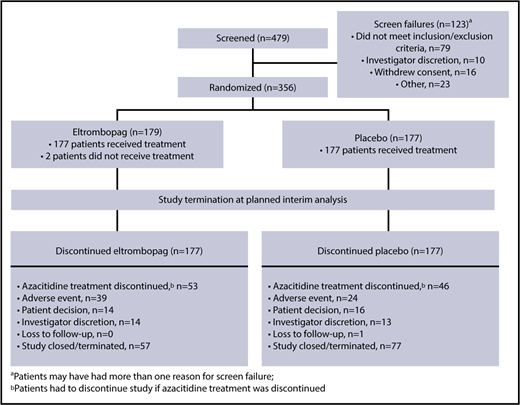
Between June 2014 and December 2015, 356 patients were enrolled in the study from 30 countries (the United States and countries in Europe, South America, and Asia), including 130 sites. Of these patients, 179 were randomized to eltrombopag (n = 64, Int-1; n = 77, Int-2; n = 38, high risk) and 177 (n = 61, Int-1; n = 83, Int-2; n = 33, high risk) to placebo. The demographics and baseline characteristics were well balanced between treatment groups (Table 1).
Patient disposition during the study is summarized in Figure 1. Based on the results of the planned interim futility analysis, the independent data monitoring committee (IDMC) recommended terminating the SUPPORT study prematurely, primarily for futility and secondarily for safety. Enrollment in the SUPPORT study had been rapid and was already complete by the time the study was terminated. Of the patients who were randomized to treatment, 2 in the eltrombopag arm did not receive treatment (Figure 1): 1 withdrew consent before the first dose and the other was withdrawn by the investigator because of stroke symptoms and Eastern Cooperative Oncology Group status 3. Efficacy analyses were conducted on the intent-to-treat (n = 356: n = 179 eltrombopag and n = 177 placebo) population; safety analyses were conducted on those who received study treatment (n = 354: n = 177 eltrombopag and n = 177 placebo). The most common reasons (≥10%) for study treatment discontinuation on eltrombopag or placebo, respectively, were study termination (32% vs 44%), azacitidine treatment discontinuation (patients had to discontinue eltrombopag/placebo if azacitidine was discontinued; 30% vs 26%), and AE (22% vs 14%). The most common reasons (≥10%) for azacitidine discontinuation in the eltrombopag and placebo arms, respectively, were study termination (35% vs 46%), AE (25% vs 19%), disease progression (including death from disease progression; 15% vs 14%), and patient decision (11% vs 9%).
Exposure to study treatments and transfusions
The median number of days on eltrombopag was less than that on placebo (83 [range, 1-477] vs 149 [range, 8-503]). The mean dose in East Asian patients was 112 (range, 60-148) mg/d in the eltrombopag arm and 122 (range, 81-147) mg/d in the placebo arm (reflecting protocol-specified dose adjustments). For non-East Asian patients, the mean dose for eltrombopag and placebo, respectively, was 205 (range, 65-293) and 245 (range, 107-316) mg/d. Average platelet counts in the eltrombopag treatment arm gradually increased through the study for patients remaining on treatment. An increasing trend was also seen in the placebo treatment arm, although the increase was not as steep. Patients in the eltrombopag arm completed fewer azacitidine cycles than those in the placebo arm (median, 4 vs 6; supplemental Figure 1, available on the Blood Web site). In total, 68 (38%) patients in the eltrombopag arm and 91 (51%) in the placebo arm received the recommended ≥6 azacitidine cycles. Many transfusions were administered during cycle 1 (51% of patients in the eltrombopag arm and 46% in the placebo arm). Transfusions were required for patients continuing to later cycles (eg, cycle 2: 41% of patients on eltrombopag and 33% on placebo) without a notable decline by treatment cycle.
Primary efficacy end point at the interim and the final analysis
Interim analysis
The IDMC recommended study termination at the planned second interim analysis (December 2015). At this time, 147 patients (eltrombopag, n = 79; placebo, n = 68) were evaluable for the primary end point. Fewer patients were platelet transfusion–independent in the eltrombopag group (n = 13/79, 16%) than in the placebo group (n = 27/68, 40%): OR, 0.25; 95% CI, 0.11-0.61; 1-sided P = 1.000 (the hypothesis was 1-sided to allow for a futility stopping rule; therefore, this result cannot be stated as statistically significant; Figure 2A). The IDMC’s recommendation to terminate the SUPPORT study prematurely was mainly the result of the primary efficacy outcome crossing the predefined futility threshold of P > .9. Secondary reasons for termination were safety related: the IDMC noted that although there was no difference in overall deaths that would indicate harm (14% eltrombopag vs 12% placebo), local review of disease progression and progression to AML indicated a trend to azacitidine/placebo advantage (14% vs 5% and 9% vs 2%, respectively) and there were higher incidence rates in serious AE and AE leading to discontinuation in the azacitidine/eltrombopag arm (60% vs 42% and 20% vs 8%, respectively). After the study was terminated, all investigators were notified of the IDMC recommendation and study procedures and eltrombopag treatment was suspended in all patients.

Primary efficacy end point. Proportion of patients who were free of platelet transfusions during cycles 1 through 4 of azacitidine therapy at (A) the interim assessment and (B) the final analysis.
Primary efficacy end point. Proportion of patients who were free of platelet transfusions during cycles 1 through 4 of azacitidine therapy at (A) the interim assessment and (B) the final analysis.
Final analyses
Primary results from the final study analysis (conducted in July 2016) performed on data from the time of study termination (n = 356) were consistent with the results of the interim analysis. Fewer patients were platelet transfusion–independent in the eltrombopag group (n = 28/179, 16%) than in the placebo group (n = 55/177, 31%): OR, 0.37; 95% CI, 0.21-0.65; 2-sided P = .001 (Figure 2B).
Secondary efficacy end points at the final analysis
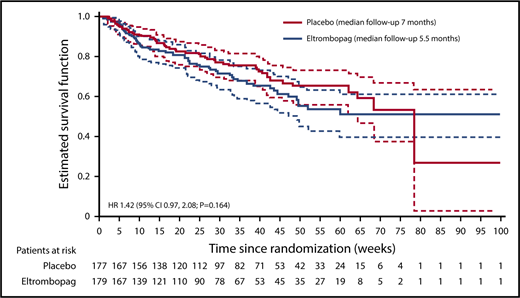
Because of hierarchical testing of end points, no formal statistical tests were performed for secondary efficacy end points. The P values provided are nominal. No multiplicity adjustment was made; therefore, statistical interpretation should be made with caution. At the final analysis, 108 patients had died: 57 patients (32%) in the eltrombopag group and 51 patients (29%) in the placebo group (hazard ratio [HR], 1.42; 95% CI, 0.97-2.08; nominal P = .164; Figure 3). In both groups, most deaths occurred within 30 days of the end of treatment: 33 patients in the eltrombopag group and 29 patients in the placebo group. The main causes of death were recorded as “disease under study” (usually disease progression; 28 patients in the eltrombopag group and 21 patients in the placebo group) and sepsis (18 and 13 patients, respectively). Other causes of death in the eltrombopag group were cardiovascular death (n = 2), hemorrhage (n = 1), other cancer (n = 1), and other noncardiovascular death (n = 7).
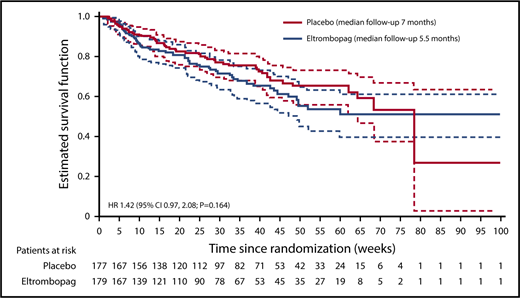
PFS, as assessed by the investigator and by central review, indicated a potentially higher risk of overall events (disease progression or death) in the eltrombopag arm (40% investigator assessment, 42% central review) compared with the placebo arm (37% investigator assessment, 38% central review); HR, 1.47; 95% CI, 1.05-2.07; nominal P = .060 for investigator assessment and HR, 1.38; 95% CI, 0.99-1.92; nominal P = .141 for central review (supplemental Table 1).
Overall response (IWG criteria, CR, marrow CR or partial remission) was reported in 20% and 35% of eltrombopag and placebo patients, respectively, by investigator assessment (OR, 0.51; 95% CI, 0.30-0.86; nominal P = .005), and CR were reported in 8% and 15% of patients (supplemental Table 2). By central review, overall responses were reported in 8% and 11% of patients (OR, 0.89; 95% CI, 0.41-1.97; nominal P = .683), and CRs were seen in 6% and 4% of eltrombopag and placebo patients, respectively (supplemental Table 2).
There was no difference in hematologic improvement in any cell lineage between the 2 treatment arms (32% for eltrombopag and 33% for placebo patients; supplemental Table 3). Median duration of platelet and neutrophil count improvement was shorter in eltrombopag recipients than in placebo recipients (platelet count: 118 days, 95% CI, 99-189, eltrombopag; 155 days, 95% CI, 134-183, placebo; neutrophil count: 110 days, 95% CI 64-150, eltrombopag; 162 days, 95% CI, 113-176, placebo).
Rate of progression to AML, confirmed by investigator assessment, tended to be higher in the eltrombopag arm compared with placebo (15% and 9%, respectively; OR, 1.59; 95%, CI 0.81-3.14; nominal P = .079, Table 2). This was also observed by central review (OR, 2.04; 95% CI, 0.90-4.65; nominal P = .042). When considering confirmation by central review or investigator assessment, 53/356 (15%) patients had progression to AML during the study: 20/177 (11%) patients in the placebo arm and 33/179 (18%) patients in the eltrombopag arm (Table 2; patient listings shown in supplemental Table 4). Nine patients evaluated as having progression to AML had >20% blasts at baseline as assessed by central review (eltrombopag n = 6; placebo n = 3), whereas none of the patients with progression to AML had >20% blasts at baseline by local review. Evaluation of patients by baseline IPSS risk category (investigator assessment) indicated that within the Int-1 category, a higher proportion of patients had progression to AML by central assessment in the eltrombopag arm compared with the placebo arm (14% vs 2%) (supplemental Table 5).
To further investigate any potential association of genetic mutations at baseline and progression to AML, we used targeted next-generation sequencing for 53 genes commonly mutated in MDS and/or AML, including a set of 18 genes that have been associated with disease progression.25-30 Overall, 211 patients (85%) had baseline whole blood samples (n = 101 eltrombopag; n = 110 placebo) and consented for translational research analysis. A subset of 11 genes (RUNX1, TET2, TP53, STAG2, SRFS2, ASXL1, NRAS, BCOR, U2AF1, DNMT3A, and SF3B1) were most frequently mutated (>5% of the total cohort at baseline in at least 1 arm) in our patient cohort. A slightly higher incidence of mutations associated with MDS or known to be of prognostic importance were observed in the placebo arm (Table 3).
Safety
At the final assessment, 354 patients were evaluable for safety; results were consistent with those seen at the interim analysis (n = 243). AE with the greatest difference between the eltrombopag and placebo arms were febrile neutropenia (31% vs 21%), neutropenia (31% vs 26%), nausea (31% vs 26%), and diarrhea (25% vs 14%) (Table 4). AE leading to permanent discontinuation of study treatment were reported in 45 patients (25%) in the eltrombopag arm compared with 24 patients (14%) in the placebo arm. Six patients died because of AE considered by the investigator (blinded to treatment) as related to study treatment (n = 5, eltrombopag [leukemia, acute myocardial infarction, acute respiratory failure, pulmonary embolism, and sepsis]; n = 1, placebo [B-cell lymphoma]).
Worst-case changes from baseline in laboratory parameters based on CTCAE grade showed a similar trend in both arms, except for hemoglobin. For hemoglobin laboratory values, an increase to CTCAE grade 3 was reported in 89 patients (51%) in the eltrombopag arm and 76 patients (43%) in the placebo arm.
Five patients presented with high liver enzyme levels and were suspected as possible cases of Hy law (alanine aminotransferase [ALT] ≥3× upper limit of normal [ULN] and bilirubin ≥2× ULN [>35% direct] or ALT ≥3× ULN and international normalized ratio >1.5, if international normalized ratio was measured); n = 4, eltrombopag; n = 1, placebo, of which 2 cases were subsequently confirmed without confounding factors. One patient (from the eltrombopag arm) met both the laboratory and medical criteria of Hy's law. Another patient (from the eltrombopag arm) met the medical but not all the laboratory criteria (ALT and aspartate aminotransferase were >3× ULN, total bilirubin was <2× ULN) of Hy's law; all values were normalized by day 80 after the first dose.
Discussion
The phase 3 SUPPORT study showed that the concomitant addition of eltrombopag to azacitidine did not meet its primary end point of improved platelet transfusion independence during cycles 1 through 4 vs placebo plus azacitidine. The planned interim analysis demonstrated that the primary efficacy end point crossed the predefined futility threshold (16% eltrombopag vs 40% placebo: OR, 0.25; 95% CI, 0.11-0.61; 1-sided P = 1.000). The IDMC noted that although there was no difference in overall deaths that would indicate harm, results from disease progression and progression to AML analyses indicated a trend toward a placebo/azacitidine advantage. Additionally, the incidence rates of serious AE and AE leading to discontinuation were higher in the eltrombopag/azacitidine arm than in the placebo/azacitidine arm. Based on these findings, the study was terminated prematurely on the recommendation of the IDMC. The final results were consistent with the results of the interim analysis.
The efficacy and safety results of this study contrast with those of recent clinical studies of eltrombopag monotherapy in patients with MDS, which showed an acceptable safety profile of eltrombopag in patients with low- and high-risk MDS and positive outcomes on thrombocytopenia.15-17,32 There were no indicators that would predict the outcome of this phase 3 trial.18,19
Preclinical and single-agent clinical studies of eltrombopag suggest that as a single agent, eltrombopag suppresses malignant myeloid blast proliferation33-36 ; hence, the findings of this trial were unexpected. One hypothesis for our findings is a potential inhibition of the effects of azacitidine by eltrombopag when given concomitantly. This question remains unanswered and is the subject of further, ongoing research. We explored the role of known molecular prognostic factors in MDS and were unable to detect imbalances between the treatment arms in this trial that would fully explain the observed clinical outcome.
An increase in blast counts could be reflective of transient stimulatory effects on specific MDS/AML clones involving BM progenitor expansion and mobilization from the marrow niche rather than true leukemic transformation/progression. Data from a phase 2 study in patients with low-risk/Int-1 MDS showed higher rates of AML progression in romiplostim- vs placebo-treated patients, which resulted in early study termination.37 However, a longer term follow-up analysis, including central review of morphology data, with a median of 27.5 months showed that survival and AML progression rates were similar between romiplostim and placebo.37 Unfortunately, the short duration of exposure to eltrombopag and termination of follow-up procedures makes assessment of causality of apparent progression difficult to determine.
In conclusion, eltrombopag in combination with azacitidine was inferior to placebo/azacitidine in this phase 3 study in patients with intermediate- and high-risk MDS and thrombocytopenia. Findings from this study do not indicate a role for combining eltrombopag with azacitidine in patients with intermediate- and high-risk MDS.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all patients and coinvestigators for their participation and contribution to this study and Rebecca Helson, of Mudskipper Business Ltd for medical editorial assistance.
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation.
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. This trial data availability is in accordance with the criteria and process described on www.clinicalstudydatarequest.com.
Authorship
Contribution: M.D., H.C., P.F., M.M., A.V., and U.P. served as investigators in this study and enrolled patients. M.S.O.P., P.B., P.M.R., and J.C. contributed to the analysis, interpretation, and reporting of the data. The first draft was written by Rebecca Helson based on discussions with and expert guidance from all authors. All authors contributed to data interpretation, reviewed and provided their comments on this manuscript, and approved the final version.
A complete list of the Study of Eltrombopag in Myelodysplastic Syndromes Receiving Azacitidine principal investigators can be found in the supplemental appendix.
Conflict-of-interest disclosure: M.D. participated in speakers’ bureaus for Celgene, Novartis Pharmaceuticals Corporation, and Janssen; received consulting fees from Novartis Pharmaceuticals Corporation, Pfizer, Janssen, and Roche; received honoraria from Novartis Pharmaceuticals Corporation, Pfizer, Janssen, and Roche; and received research grants from Celgene, and Janssen. H.C. received research funding from GlaxoSmithKline and Pfizer; participated in advisory boards for Abbvie, Amgen, Janssen Cilag, Roche, Gilead, GlaxoSmithKline, Novartis, and Sanofi; and served as a medical advisor for Wyeth Pharmaceuticals. P.F. received research funding from Celgene, Janssen, Novartis Pharmaceuticals Corporation, Astex, and Teva and received honoraria from Celgene, Novartis Pharmaceuticals Corporation, and Teva. M.M. received research funding from Amgen, Celgene, GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corporation, and Roche and participated in speakers’ bureaus for Celgene, Johnson & Johnson. M.S.O.P., P.B., P.M.R., and J.C. are employed by Novartis Pharmaceuticals Corporation. U.P. received honoraria from Amgen, Celgene, GlaxoSmithKline, and Novartis Pharmaceuticals Corporation and received research funding from GlaxoSmithKline. A.V. declares no competing financial interests.
Correspondence: Michael Dickinson, Clinical Haematology, Peter MacCallum Cancer Centre and Royal Melbourne Hospital, Melbourne, VI 3000, Australia; e-mail: michael.dickinson@petermac.org.