In this issue of Blood, use a high-throughput exome-sequencing approach to identify 2 biallelic loss-of-function mutations in PTPRJ that caused autosomal-recessive thrombocytopenia and a bleeding disorder in 2 siblings.1
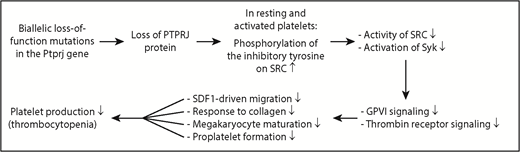
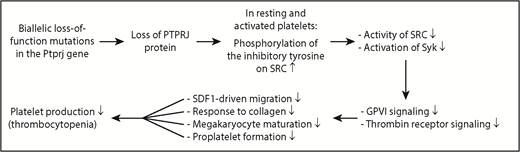
Diagram showing the mechanism underlying IT in humans caused by 2 biallelic loss-of-function mutations in the Ptprj gene. Loss of PTPRJ protein caused by biallelic mutations of Ptprj increases phosphorylation of the inhibitory tyrosine on the SRC kinase; reduces activation of SRC and its downstream signaling molecule Syk; weakens GPVI- and thrombin receptor-mediated signaling; impairs SDF1-driven migration of MKs, collagen-induced platelet response, terminal MK maturation, and proplatelet formation; and ultimately results in thrombocytopenia.
Diagram showing the mechanism underlying IT in humans caused by 2 biallelic loss-of-function mutations in the Ptprj gene. Loss of PTPRJ protein caused by biallelic mutations of Ptprj increases phosphorylation of the inhibitory tyrosine on the SRC kinase; reduces activation of SRC and its downstream signaling molecule Syk; weakens GPVI- and thrombin receptor-mediated signaling; impairs SDF1-driven migration of MKs, collagen-induced platelet response, terminal MK maturation, and proplatelet formation; and ultimately results in thrombocytopenia.
Inherited thrombocytopenia (IT) is an extremely heterogeneous group of thrombocytopenic conditions. Classification of IT based on the inheritance pattern or clinical symptoms other than thrombocytopenia is not always reliable due to the high frequency of sporadic cases with de novo gene mutations, partial penetrance of the mutations, and variable modes of presentation in patients with the same gene mutations. Classification of IT based on platelet size can be helpful and relatively reliable.2 However, characterization of clinical and laboratory findings that correlate with an identified genetic abnormality is essential to define a particular IT as a specific disease entity.
ITs have various phenotypes and are caused by mutations in many different genes. The genetic defects responsible for an IT were first defined in 2 conditions: Bernard-Soulier syndrome (BSS) and Wiskott-Aldrich syndrome (WAS). BSS typically is associated with a severe bleeding tendency and is caused by mutations of genes encoding the components of glycoprotein (GP) complex GPIb/IX/V.3 WAS is associated with bleeding, immune deficiency, allergy, autoimmunity, and malignancy, and is due to mutations in the WAS gene.4 In recent years, application of high-throughput sequencing, especially whole-exome and whole-genome sequencing, had led to identification of many genes responsible for ITs.5 Mutation of the myeloproliferative leukemia (MPL) receptor impairs the maintenance of the hematopoietic stem cell (HSC) compartment and HSC differentiation into megakaryocytes (MKs). Mutations of transcription factors critical for megakaryopoiesis, such as RUNX1, GATA1, and others, alter the expression of numerous genes and impede MK maturation. Mutations of components of the GPIIb/IIIa complex, actomyosin cytoskeleton, or microtubules, such as ITGA2B, MYH9, and others, block proplatelet formation from mature MKs and/or conversion of proplatelets to platelets. The newer findings markedly advance our understanding of the molecular pathogenesis of ITs and the physiologic mechanisms of megakaryopoiesis and platelet biogenesis. Despite this progress, causative mutations have been identified for only ∼50% of ITs; so much is still to be learned from studies of patients falling into this disease category.
PTPRJ encodes the receptor-type protein tyrosine phosphatase CD148 (RPTP). It is the most abundant RPTP in platelets and MKs, but is expressed in other cell types, both hematopoietic and nonhematopoietic. PTPRJ contains a single intracellular catalytic phosphatase domain, a transmembrane domain, and 9 heavily glycosylated extracellular fibronectin type III–like repeats.6 PTPRJ dephosphorylates the C-terminal inhibitory tyrosine residues of the Src family of protein tyrosine kinases (SFKs), thus enhancing SFK activity.7 Functional deficiency of PTPRJ in mouse platelets reduces SFK activity in both resting and GPVI- or GPIIb/IIIa-activated platelets, and decreases GPVI expression and GPVI-induced activation of FcRγ chain, Syk and phospholipase Cγ2.7 Consequently, PTPRJ-deficient mouse platelets exhibit an impairment of spreading on collagen, fibrinogen, and fibronectin; GPVI-mediated aggregation and secretion; and stromal cell–derived factor (SDF)-driven migration on fibronectin.7 Nonetheless, PTPRJ-deficient mice display only slightly impaired hemostasis following tail excision and have only a mild bleeding diathesis and largely normal number of platelets.7
In this issue of Blood, Marconi et al use high-throughput sequencing to identify 2 biallelic loss-of-function mutations in the PTPRJ gene as a new cause of IT in humans (see figure). Both mutations result in frameshift and insertion of a premature stop codon, messenger RNA decay, and almost no production of PTPRJ protein. The nearly complete loss of PTPRJ caused by biallelic loss-of-function mutations is transmitted as a recessive trait. Most IT forms are characterized by large platelets whereas the patients with PTPRJ deletion possess small platelets. The other prototypical IT forms that present as a recessive and nonsyndromic thrombocytopenia with normal or small platelets are congenital amegakaryocytic thrombocytopenia caused by MPL or thrombopoietin mutations, and WAS and its variant X-linked thrombocytopenia caused by WAS mutations.2 Diagnosis of ITs with small platelets should consider PTPRJ mutations, although the underlying mechanism is not clear.
Platelets from the PTPRJ-deficient patients studied by Marconi et al display impaired activation by the GPVI-specific agonist convulxin and the thrombin receptor–activating peptide but normal responsiveness to adenosine 5′-diphosphate. The defects observed are the result of impaired signal transduction from the receptor, evidenced by a marked inhibition of convulxin-induced protein tyrosine phosphorylation. PTPRJ deficiency leads to a constitutive increase in phosphorylation of the inhibitory tyrosine on SRC kinase in resting and convulxin-activated human platelets, resulting in reduced activation of SRC. Consistently, the activation of the downstream tyrosine kinase Syk in response to convulxin is markedly reduced. Normally, PTPRJ regulates phosphorylation of the inhibitory tyrosine residues on SRC and maintains a baseline level of SRC activation, thus functioning as a universal signal regulator of human platelet activation. It is conceivable that PTPRJ-deficiency-caused impairment of SFK activation is responsible for many of the signaling defects observed in human platelets and MKs.
What is the underlying mechanism by which the deletion of PTPRJ induces thrombocytopenia in humans? Marconi et al show that PTPRJ deficiency in the 2 patients studied impairs SDF1-driven migration of MKs on type I collagen or fibrinogen; reduces the platelet activation response to collagen, convulxin, and TRAP; hampers terminal MK maturation; and decreases proplatelet formation, all of which may contribute to the underlying thrombocytopenia. In the bone marrow, MKs migrate from the osteoblastic niche to sinusoid-enriched areas to protrude proplatelets through vascular endothelium into the blood stream. This migration is driven by SDF1 and is critical for platelet production.8 Defects in SDF1-driven migration are associated with human IT.9 However, PTPRJ-deficient mice have defective SDF1-driven migration, but display largely normal numbers of platelets.7 The cause of this discrepancy between humans and mice is not well understood. Defective proplatelet branching can also impair platelet production and is directly associated with the pathogenesis of human IT.10 Both reduced activation of SFKs and defective MK maturation can cause the defective proplatelet formation,7 thus contributing to the pathogenesis of human thrombocytopenia. As with many “experiments of nature,” recognition of PTPRJ as a new IT gene advances our understanding of the molecular mechanisms underlying megakaryopoiesis and platelet biogenesis in humans.
Conflict-of-interest disclosure: The authors declare no competing financial interests.