Abstract
Relapse of the original disease is a major cause of death after allogeneic hematopoietic cell transplantation for acute leukemias. There is growing evidence that relapses may be explained not only by resistance to chemotherapy but also by the escape of tumor cells from the control of the allogeneic immune response. Mechanisms of immune evasion can involve abrogation of leukemia cell recognition due to loss of HLA genes, immunosuppression by immune-checkpoint ligand expression, production of anti-inflammatory factors, release of metabolically active enzymes, loss of proinflammatory cytokine production, and acquisition of novel driver mutations that promote leukemia outgrowth. These mechanisms, and therapeutic targeting of immune escape, will be discussed. We divide the evidence in support of immune-escape mechanisms into animal studies, human laboratory studies, and human clinical experience. A better understanding of the molecular pathways connected to immune escape and relapse may help to improve our therapeutic armamentarium against acute myeloid leukemia relapse.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 1386.
Disclosures
Editor Nancy Berliner, CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, and the authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe impaired leukemia cell recognition and inhibitory immune checkpoint molecules as mechanisms of escape of tumor cells from the control of the allogeneic immune response after allogeneic hematopoietic cell transplantation (allo-HCT) for acute leukemias, according to a review
Determine other mechanisms of escape of tumor cells from the control of the allogeneic immune response after allo-HCT for acute leukemias, according to a review
Explain selected therapeutic strategies against immune escape in acute myeloid leukemia relapse after allo-HCT, according to a review
Release date: March 21, 2019; Expiration date: March 21, 2020
Introduction
For a majority of the patients with acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), allogeneic hematopoietic cell transplantation (allo-HCT) is the only curative treatment. Early after allo-HCT, infections and acute graft-versus-host disease (GVHD)1 are major causes of death.2 After day 100 post-allo-HCT, relapse is the major cause of death and its incidence and outcome have not significantly improved over the last decades.2,3 Relapse means that leukemia that had been in remission recurs on the molecular level (molecular relapse) or on the hematological level (hematological relapse). Although a number of factors come into play, including chemoresistance to the conditioning regimen, in many cases, relapse indicates that initially the T cells were able to control the disease but later the leukemia cells have managed to escape from this control.
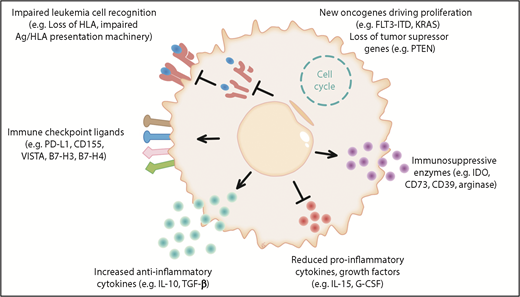
Novel cellular therapies, such as chimeric antigen receptor T cells, have shown very promising results in lymphoblastic leukemia; however, relapse remains a major clinical challenge and, therefore, a better understanding of the biology of relapse will be fundamental in the upcoming new era of advanced cell therapies. Different experimentally proven and hypothetical mechanisms of immune evasion post allo-HCT are shown in Figure 1.
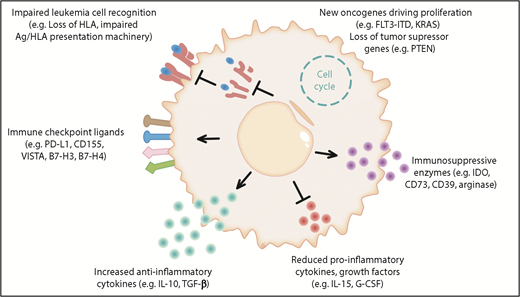
The different mechanisms of immune evasion post allo-HCT are shown in 6 major categories. (1) Impaired leukemia cell recognition via genomic loss/impaired expression of HLA or defects in the antigen (Ag)/HLA-presentation machinery.11,84 (2) Upregulation of immune-checkpoint molecules: a role for programmed death 1 (PD-1) is shown for Hodgkin lymphoma and the B7 family member/CTLA4 axis is shown in clinical trials; hypothetically, CD155, VISTA, B7-H3, B7-H4, and others could play a role in AML immune escape post allo-HCT.28,85 (3) Increased anti-inflammatory cytokine production was shown in leukemia cells, which could hypothetically play a role in relapse; examples are interleukin-10 (IL-10) and transforming growth factor β (TGF-β). (4) Reduced production of IL-15 was shown to lower anti-AML immune responses post allo-HCT.41 Hypothetically, suppression of other cytokines such as granulocyte colony-stimulating factor (G-CSF), which promotes Ag-presenting cell (APC) maturation, could play a role in AML immune escape post allo-HCT. (5) Production of the metabolic active molecules CD73, indoleamine 2,3-dioxygenase (IDO), and arginase by leukemia cells was shown and CD73 deletion or inhibition promoted graft-versus-leukemia (GVL) effects in the mouse model.51 These molecules could play a role in AML immune escape. (6) Acquisition of novel oncogenic mutations and loss of tumor-suppressor genes post allo-HCT have been described.7,52-54 Hypothetically, this increased proliferation could promote immune escape as leukemia cells outnumber T cells and NK cells. FLT3-ITD, FLT3–internal tandem duplication; PD-L1, programmed death ligand 1.
The different mechanisms of immune evasion post allo-HCT are shown in 6 major categories. (1) Impaired leukemia cell recognition via genomic loss/impaired expression of HLA or defects in the antigen (Ag)/HLA-presentation machinery.11,84 (2) Upregulation of immune-checkpoint molecules: a role for programmed death 1 (PD-1) is shown for Hodgkin lymphoma and the B7 family member/CTLA4 axis is shown in clinical trials; hypothetically, CD155, VISTA, B7-H3, B7-H4, and others could play a role in AML immune escape post allo-HCT.28,85 (3) Increased anti-inflammatory cytokine production was shown in leukemia cells, which could hypothetically play a role in relapse; examples are interleukin-10 (IL-10) and transforming growth factor β (TGF-β). (4) Reduced production of IL-15 was shown to lower anti-AML immune responses post allo-HCT.41 Hypothetically, suppression of other cytokines such as granulocyte colony-stimulating factor (G-CSF), which promotes Ag-presenting cell (APC) maturation, could play a role in AML immune escape post allo-HCT. (5) Production of the metabolic active molecules CD73, indoleamine 2,3-dioxygenase (IDO), and arginase by leukemia cells was shown and CD73 deletion or inhibition promoted graft-versus-leukemia (GVL) effects in the mouse model.51 These molecules could play a role in AML immune escape. (6) Acquisition of novel oncogenic mutations and loss of tumor-suppressor genes post allo-HCT have been described.7,52-54 Hypothetically, this increased proliferation could promote immune escape as leukemia cells outnumber T cells and NK cells. FLT3-ITD, FLT3–internal tandem duplication; PD-L1, programmed death ligand 1.
Impaired leukemia cell recognition
Elimination of leukemia cells post allo-HCT relies mainly on their recognition by T cells and natural killer (NK) cells. T cells recognize as targets immunogenic peptides presented by self-HLA molecules and also structural epitopes of non-self-HLAs, whereas NK cells are activated in response to the absence or downregulation of self-HLA class I molecules on tumor cells.
Human laboratory studies and human clinical experience show that the loss of the mismatched HLA in the genome of the leukemia represents an important immune-escape mechanism that allows the leukemia to relapse after allo-HCT.4 Genomic loss of mismatched HLA occurs through a mechanism of copy-neutral loss of heterozygosity, eliminating the incompatible HLA alleles without decreasing the overall level of expression of HLA class I molecules. This explains why an NK-cell–mediated response is not activated. Several studies have investigated the incidence of HLA loss after haploidentical allo-HCT, reporting that it can account for up to one-third of relapses in this setting, irrespective of the strategy used to control T-cell alloreactivity and prevent GVHD.5,6 There have also been reports of cases of genomic HLA loss after allo-HCT from unrelated donors,7,8 but larger studies are warranted to better appreciate the frequency and clinical relevance of the phenomenon in this setting. This will be facilitated by the recent development of straightforward polymerase chain reaction (PCR)-based methods to detect HLA loss, allowing routine diagnostics for these relapse variants.9 Figure 1 outlines HLA loss and the other mechanisms of posttransplantation immune evasion. Recent studies based on patient samples show that downregulation of HLA class II molecules and its regulators (class II major histocompatibility complex [MHC-II] transactivator [CIITA], IFI30, HLA-DMA, HLA-DMB, and CD74) can drive leukemia relapse after both HLA-matched and -mismatched transplants.10,11
Studies performed in the absence of allo-HCT show a role of downregulation of HLA class II molecules in hematological malignancies as a mechanism of immune evasion. A recent study showed that chronic myeloid leukemia (CML) cells actively evade host immune surveillance through cytokine-mediated downregulation of HLA class II expression.12 Both Hodgkin lymphoma and aggressive B-cell lymphomas13,14 display HLA class II downregulation, which limits the host antitumor immune response. In agreement with this, higher HLA class II expression on tumor cells correlated with better prognosis and response to immunotherapy.13,15,16
Inhibitory immune-checkpoint molecules
Besides loss of HLA leading to less alloantigen recognition, other immune-escape mechanisms may promote AML relapse, however, these are less well studied. A mechanism that is mainly supported by clinical trial data is the role of inhibitory immune-checkpoint molecules for AML relapse.
In the nontransplant setting, a recent clinical study on anti–programmed death 1 (PD-1) treatment of refractory or relapsed AML showed efficacy in a nonrandomized, open-label, phase 2 study.17
In the allo-HCT, a role for PD-1 was shown for Hodgkin lymphoma because patients relapsing with this disease showed promising responses when treated with anti–PD-1 antibodies.18 Blocking CTLA4 in AML relapse was shown to be effective in clinical trials.19-21
Moreover, recent studies on patient samples demonstrated the deregulated expression of immune-regulatory molecules including programmed death ligand 1 (PD-L1), B7-H3, and PVRL2 (CD155) on AML cells at relapse post allo-HCT (Figure 1).10
Myeloproliferative neoplasms (MPNs) that develop into AML often lose their JAK JAK2V617F mutation. MPNs are potentially sensitive to immunotherapy as they respond to interferon-α treatment22 and because JAK2-specific23 and Calreticulin-specific24 T cells have been reported. Recently, it was shown that oncogenic JAK2V617F activity caused STAT3 and STAT5 phosphorylation, which enhanced PD-L1 promoter activity and PD-L1 protein expression.25 PD-L1 surface expression on JAK2V617F-mutant cells reduced metabolic activity and cell-cycle progression of T cells that were in contact with these myeloid cells.23 These findings link oncogenic JAK2V617F signaling to immune escape in the nontransplant setting.23
Another category of diseases that can affect antitumor responses in the nontransplant setting is Myc-driven lymphomas, as it was shown that Myc increases the expression of CD47 and PD-L1.26 CD47 binds to signal regulator protein-α on macrophages and dendritic cells and thereby blocks antigen uptake.27 PD-L1 and CD47 transcription was controlled directly by MYC protein, and restoring CD47 or PD-L1 expression by transfection reversed the phenotype.26 The authors showed that inactivation of the Myc gene led to markedly decreased levels of immunosuppressive PD-L1, and the leukocyte surface antigen CD47, which limits activation of antigen-presenting cells (APCs).26
How important PD-L1 expression on leukemia or lymphoma cells post allo-HCT is for immune escape is currently unclear. Retrospective studies on patients treated with anti–PD-1 for Hodgkin lymphoma relapse post allo-HCT indicate not only efficacy but also high GVHD rates, which is a potential limitation of immune-checkpoint inhibition in the post allo-HCT setting.18
In addition to the expression of inhibitory ligands on malignant cells, changes affecting the donor-derived immune effectors might be of fundamental relevance in explaining posttransplantation relapse. In the allo-HCT setting, a recent study documented the coexpression of multiple inhibitory checkpoints, including PD-1, TIGIT, and KLRG-1, on minor histocompatibility antigen-specific T cells harvested from patients at the time of relapse.28 Moreover, these changes appear to be even more profound when examining the bone marrow compartment of transplanted patients, with exhausted central memory and memory stem T cells infiltrating the site where interactions between leukemia and T cells are expected to occur.29
Modulation of anti- and proinflammatory cytokine production
The production of anti-inflammatory cytokines can paralyze immune responses. Increased anti-inflammatory cytokine production was shown in different types of leukemia cells.30 A theoretical mechanism of immune escape post allo-HCT is the production of anti-inflammatory cytokines by AML cells.
Studies performed in the absence of allo-HCT indicated that CML cells produce transforming growth factor (TGF-β),30 which antagonizes the CIITA/MHC-II axis31 thereby rendering leukemia cells less immunogenic. Interleukin-4 (IL-4) was also produced by CML cells12 and comparable to TGF-β, IL-4 reduces MHC-II expression.32 Immunosuppressive IL-10 was shown to be produced by chronic lymphocytic leukemia (CLL) cells, which causes immune evasion.33 IL-4 and IL-10 were also detected in AML cells.34
Conversely, proinflammatory cytokines, which are normally produced by healthy myeloid or lymphoid progenitors, could be disadvantageous for the leukemia cells and therefore it would be reasonable for the malignant cell to block their production. Examples of proinflammatory cytokines produced by myeloid progenitors are granulocyte colony-stimulating factor (G-CSF), IL-15, and interferon-γ (IFN-γ).
A clinical study performed in the absence of allo-HCT showed that in B-lineage ALL, low IFN-γ expression was connected to high-risk groups suggesting that a defect in IFN-γ production allows the leukemia cells to escape immunosurveillance.35
IL-15 is normally strongly produced by myeloid progenitors36 and, at the same time, is a cytokine that promotes leukemia control. Different studies have used the injection of exogenous IL-15 to boost the alloimmune effect37 or to expand NK cells to eliminate leukemia post allo-HCT.38 IL-15 instructs the generation of human memory stem T cells from naive precursors.39 Low serum IL-15 levels were reported to be connected to relapse after allo-HCT.40 A recent multicenter phase 1 trial evaluated the IL-15 superagonist complex ALT-803 in patients who relapsed >60 days after allo-HCT.38 The authors observed that ALT-803 led to the activation and expansion of CD8+ T cells NK cells and responses in 19% of evaluable patients.38
Therefore, high IL-15 levels in the microenvironment are unfavorable for the leukemia cells as they expand and activate effector T cells and NK cells. In the absence of allo-HCT, healthy myeloid progenitor cells produce high amounts of the proinflammatory cytokine IL-15.36 In contrast, IL-15 production is blocked in AML cells with an internal tandem duplication (ITD) of the FLT3 tyrosine kinase (FLT3-ITD).41 Animal studies performed in the allo-HCT setting showed that FLT3 inhibition along with T-cell transfer caused elimination of leukemia cells and long-term disease control. Pharmacological FLT3 inhibition caused a reduction of the expression of the transcription factor ATF4, thereby inhibiting the interferon regulatory factor 7 (IRF7) activation block. Counteracting IRF7 inhibition caused IL-15 transcription.41 The increased IL-15 levels caused major changes in T-cell metabolism. Human laboratory studies indicated that mitochondrial spare respiratory capacity and glycolytic capacity of the transferred CD8+ T cells increased upon FLT3 inhibition indicating metabolic reprogramming of leukemia-reactive T cells.41 These findings are in agreement with previous reports showing that IL-15 can lead to mitochondrial reprogramming thereby enhancing antitumor immunity.42,43
A hypothetical mechanism of immune escape is the suppression of other proinflammatory growth factors such as G-CSF, which promotes APC maturation, or cytokines such as IL-1β potentially playing a role in AML immune escape post allo-HCT (Figure 1).
Enzymes that mediate immunosuppression
Production of the metabolic active molecules indoleamine 2,3-dioxygenase-1 (IDO1),44 arginase,45 CD39,46 and CD7347 by leukemia cells in the nontransplant setting was shown. IDO is an intracellular heme-containing enzyme that catalyzes the initial, rate-limiting step in tryptophan degradation leading to kynurenine production.48 Kynurenine inhibits T-cell functions and reprograms T regulatory cells (Tregs).48 Additionally, the lack of tryptophan affects T-cell function.48 IDO1 expression by leukemia cells was correlated with an unfavorable prognosis of childhood AML in the nontransplant setting.44
Arginase leads to depletion of the amino acid arginine, which is required for T-cell function. AML cells exhibit an arginase-dependent ability to inhibit T-cell proliferation and to polarize surrounding monocytes toward a suppressive M2-like phenotype.45 In agreement with a role for soluble inhibitory factors secreted by leukemia cells, it has been shown that AML cell supernatants reduced T cells and NK-cell proliferation in the nontransplant setting.49,50
CD73 is an ectonucleotidase that leads to the production of adenosine from adenosine monophosphate (AMP). Adenosine was shown to be immunosuppressive and, therefore, inhibition of CD73 could be a therapeutic strategy to enhance leukemia control. In mouse studies in the allo-HCT setting, genetic deficiency of CD73 enhanced leukemia cell rejection after allo-HCT.51
CD39 is an ectonucleoside triphosphate diphosphohydrolase-1 that metabolized adenosine triphosphate to adenosine diphosphate and AMP and was shown to promote an immunosuppressive microenvironment in AML in the nontransplant setting.46
These enzymes involved in metabolism could play a role in leukemia and lymphoma immune escape post allo-HCT, however, this has so far not been proven.
Novel oncogenic mutations
Acquisition of novel oncogenic mutations and loss of tumor-suppressor genes post allo-HCT has been described.52 Sequencing studies have revealed that most leukemias consist of multiple genetic clones including a founding clone and subclones. Myelodysplastic syndrome (MDS) progression after allo-HCT was connected to the expansion and evolution of leukemic subclones.52 A prognostic role for different mutations after allo-HCT for MDS has been reported.53 Additionally, for AML, the pairwise comparisons of chromosomal alterations and mutational profile between leukemia samples collected at diagnosis and at posttransplantation relapse revealed frequent changes in the genomic asset, including appearance at relapse of new mutations in TET2, WT1, and RAS.7,54
Hypothetically, the increased proliferation caused by more driver mutations could promote immune escape as leukemia cells outnumber the leukemia-reactive T cells and NK cells. This concept has not been proven but the observation that novel mutations that confer a strong proliferative stimulus are found in relapsing leukemia cells points toward this possibility.
We speculate that the 6 main areas of immune escape that we describe in Figure 1 should be analyzed in a prospective trial on a defined high-risk AML patient cohort in order to better understand the biology of relapse.
Selected therapeutic strategies against immune escape in AML relapse
The many distinct mechanisms of immune evasion described in the previous paragraphs have direct and profound translational implications: if in fact most of the therapeutic approaches tested to date for relapsed patients have shown limited or negligible results, it can be envisaged that they might be far more effective when used in a selected group of patients based on a specific rationales, warranting in-depth analysis of relapsed disease to inform treatment selection.
An example in such a sense is provided by relapses with genomic loss of HLA, which currently represent a clear counterindication to the infusion of lymphocytes from the original donor (that would prove far less effective against the leukemia while retaining their potential to cause GVHD),55 also providing a criterion for the selection of a second donor in case of retransplantation.56,57
With the emergence of a number of new promising therapeutics against hematological malignancies, which agents synergize with the graft-versus-leukemia effect of allo-HCT and which others antagonize it should also be carefully considered. A remarkable example of approaches to counteract the cooperation of oncogene-driven proliferation and immune escape is provided by the combined inhibition of the driving signaling pathway and immunotherapy tested in FLT3-ITD mutant AML relapsing after allo-HCT. Retrospective studies in patients treated with sorafenib for FLT3-ITD AML relapsed post allo-HCT reported impressive responses.58-61 Although some patients seemed to benefit from sorafenib alone, other studies reported combinations of sorafenib in combination with donor lymphocyte infusion (DLI) or azacytidine.62 A retrospective analysis of 409 patients relapsing with FLT3-ITD AML after allo-HCT suggested that relapse treatment with sorafenib and DLI was superior to DLI alone.41
Preclinical studies showed that sorafenib may enhance immunogenicity of leukemia cells via induction of IL-15 production by the leukemia cells, thus enhancing T-cell activation.41 The immunological effects were also seen when midostaurin or crenolanib were used instead of sorafenib. FLT3-ITD is associated with activating transcription factor 4 induction, which inhibits IL-15 production. Tyrosine kinase inhibitor–based inhibition of activating transcription factor 4 reverts this and allows for IL-15 production.41
Therapeutic approaches that aim at activating the allogeneic immune response involve immunomodulatory drugs such as lenalidomide or panobinostat. Clinical case studies reported that treatment with lenalidomide can eliminate leukemia cells in relapsed AML after allo-HCT, however, GVHD was a serious life-threatening side effect in the reported patients.63
Other strategies to overcome immune escape post allo-HCT are the blockade of inhibitory immune checkpoints on AML cells. Feasibility of the blockade of CTLA-4 was shown in case studies including different hematological malignancies.19,20 Later, a phase 1/1b multicenter study in which 28 patients with different hematological malignancies (including AML and MDS) who relapsed after allo-HCT were enrolled analyzed the safety and efficacy of the anti-CTLA-4 antibody ipilimumab.21 The overall response rate in these patients was 32% including 23% complete remissions (CRs), and CR occurred in 4 patients with extramedullary AML.21 Responses were long-lasting as 4 patients had a durable response for over 1 year. GVHD was reported in 14% and overall immune-related side effects were reported in 21% of the patients.21 Tissue analysis showed that cytotoxic CD8+ T-cell numbers were increased in responders whereas Tregs decreased.
Additionally, anti-PD1 immunotherapy was given to patients relapsing with AML after allo-HCT; a small case series reported efficacy in some patients with limited GVHD.64 Other studies using higher anti-PD-1 dosages and including different hematological malignancies reported severe acute and chronic GVHD rates up to 55%.65 Further studies on the use of ipilimumab or anti-PD-1/PD-L1 in AML/MDS relapse after allo-HCT are currently ongoing to determine the value of this approach to overcome immune escape post allo-HCT.66
Cellular therapy in the form of preemptive DLI was reported in retrospective analyses showing efficacy to prevent hematological relapse.55 One prospective study and several retrospective studies reported a response of overt AML/MDS relapse to therapeutic DLI.67-69 Several studies suggest that DLIs in the setting of molecular or cytogenetic relapse, that is, preemptive DLI results in a higher response rate than DLI for overt relapse.70 Other studies have analyzed the combination of DLI with chemotherapy71 or DLI and hypomethylating agents.72,73 A study on the activity of azacytidine in 181 patients who relapsed after an allograft for AML (n = 116) or MDSs (n = 65) showed that the concurrent administration of DLI did not improve either response rates or overall survival in patients treated with azacytidine.74 Conversely, a phase 1 study was conducted in patients with AML relapse with the following scheme: chemotherapy followed by DLI followed by azacytidine showed clinical benefit in a fraction of patients.75 This approach may help overcome immune escape because inhibition of DNA methylation can activate endogenous retroviral expression and double stranded RNA in cancer cells.76,77 These retroviruses then cause stimulation of Toll-like receptor signaling, which induces an antiviral interferon response that enhances antitumor immunity.76,77 This concept is supported by in vitro studies reporting that cancer/testis antigens were increasingly expressed in AML cell lines treated with 5-aza-2′-deoxycytidine.78,79 A prospective preemptive phase 2 study showed that treatment with 5-azacytidine in patients with a decrease of the CD34 donor chimerism or an increase of minimal residual disease (MRD) induced an increase of the donor chimerism or a decline of MRD, respectively.80 The efficacy of the combination of azacytidine and DLI for hematological relapse after allo-HCT has been shown in different independent studies including 1 phase 1 study,75 a retrospective analysis of the German Cooperative Transplant Study Group,73 a retrospective single-center analysis,81 and others.72,82 These therapies can induce long-lasting remissions in a fraction of the patients who relapse after allo-HCT. One study reported an overall response rate of 33% including 27% CRs and the overall survival at 2 years was 29%.73 In a retrospective single-center analysis the authors reported that intensive chemotherapy yielded better overall survival and progression-free survival than hypomethylating agents when combined with DLI.83
Acknowledgments
The authors thank Cristina Toffalori for her help in outlining the figure.
This work was supported by a European Research Council (ERC) Consolidator grant (681012 GVHDCure [R.Z.]); the Deutsche Forschungsgemeinschaft (Germany) for SFB1160 (R.Z.), ZE 872/4-1, and TRR167 (B6 [R.Z.]); the Italian Ministry of Health (RF-2011-02351998 and JTC2012 TRANSCAN HLALOSS [L.V.]); the Associazione Italiana per la Ricerca sul Cancro (Start-Up Grant #14162 [L.V.]); and the Deutsche Knochenmarkspenderdatei (DKMS) Mechtild Harf Foundation (DKMS Mechtild Harf research grant 2015 [L.V.]).
Authorship
Contribution: R.Z. and L.V. performed literature research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert Zeiser, Department of Hematology, Oncology and Stem Cell Transplantation, Freiburg University Medical Center, Hugstetter Str 55, 79106 Freiburg, Germany; e-mail: robert.zeiser@uniklinik-freiburg.de; and Luca Vago, Unit of Immunogenetics, Leukemia Genomics and Immunobiology and Hematology and Bone Marrow Transplantation Unit, San Raffaele Scientific Institute, via Olgettina, 60 Milan, Italy; e-mail: vago.luca@hsr.it.