

Key Points
A high percentage of patients with β-thalassemia had improvement in hemoglobin or transfusion burden after receiving luspatercept.
Findings support a randomized clinical trial to assess the efficacy and safety of luspatercept for treatment of β-thalassemia.

Abstract
β-thalassemia is a hereditary disorder with limited approved treatment options; patients experience anemia and its complications, including iron overload. The study aim was to determine whether luspatercept could improve anemia and disease complications in patients with β-thalassemia. This open-label, nonrandomized, uncontrolled study consisted of a 24-week dose-finding and expansion stage (initial stage) and a 5-year extension stage, currently ongoing. Sixty-four patients were enrolled; 33 were non–transfusion dependent (mean hemoglobin, <10.0 g/dL; <4 red blood cell [RBC] units transfused per 8 weeks), and 31 were transfusion dependent (≥4 RBC units per 8 weeks). Patients received 0.2 to 1.25 mg/kg luspatercept subcutaneously every 21 days for ≥5 cycles (dose-finding stage) and 0.8 to 1.25 mg/kg (expansion cohort and 5-year extension). The primary end point was erythroid response, defined as hemoglobin increase of ≥1.5 g/dL from baseline for ≥14 consecutive days (without RBC transfusions) for non–transfusion-dependent patients or RBC transfusion burden reduction ≥20% over a 12-week period vs the 12 weeks before treatment for transfusion-dependent patients. Eighteen non–transfusion-dependent patients (58%) receiving higher dose levels of luspatercept (0.6-1.25 mg/kg) achieved mean hemoglobin increase ≥1.5 g/dL over ≥14 days vs baseline. Twenty-six (81%) transfusion-dependent patients achieved ≥20% reduction in RBC transfusion burden. The most common grade 1 to 2 adverse events were bone pain, headache, and myalgia. As of the cutoff, 33 patients remain on study. In this study, a high percentage of β-thalassemia patients receiving luspatercept had hemoglobin or transfusion burden improvements. These findings support a randomized clinical trial to assess efficacy and safety. This study was registered at www.clinicaltrials.gov as #NCT01749540 and #NCT02268409.
Introduction
β-thalassemia is a hereditary red blood cell (RBC) disorder caused by mutations in the β-globin gene.1 These mutations lead to oxidative stress and premature death of erythroblasts, resulting in ineffective erythropoiesis and erythroid expansion in the bone marrow.2,3 Comorbidities, including bone deformities, splenomegaly, and leg ulcers, are the result of 1 or more factors associated with the disease, including ineffective erythropoiesis, anemia, and iron overload.1
Patients with β-thalassemia major have a severe form of the disease and are generally transfusion dependent. However, receiving regular RBC transfusions leads to iron overload, necessitating daily iron chelation therapy.4 β-thalassemia intermedia includes less severe forms of the disease that may be initially non–transfusion dependent but over time may become transfusion dependent. Although these patients are generally non–transfusion dependent, iron overload can result from increased intestinal iron absorption because of reduced hepcidin levels.5 Sporadic or even regular transfusions may therefore be required.4,6,7
Current treatment options for β-thalassemia are mostly limited to supportive therapy. Hematopoietic stem cell transplantation is currently the only possible curative treatment; however, it is only suitable for a small percentage of patients.8 Gene therapy clinical trials for β-thalassemia are in the early stages.9 The use of erythropoiesis-stimulating agents, such as erythropoietin, in β-thalassemia is not supported by controlled clinical trials.10,11
Members of the transforming growth factor β superfamily of ligands, including growth differentiation factors and activins, have been shown to act as inhibitors of late-stage erythropoiesis.12 Luspatercept (ACE-536) is a novel recombinant fusion protein that acts as a trap for these ligands. In healthy volunteers, luspatercept was well tolerated and associated with dose-dependent increases in hemoglobin levels.13 RAP-536, a murine analog of luspatercept, has also been shown to reduce oxidative stress and anemia in a mouse model of β-thalassemia.14
The aim of this phase 2 study was to determine a tolerable and active dose level and schedule of luspatercept in adults with transfusion-dependent or non–transfusion-dependent β-thalassemia.
Methods
Study oversight
The study was approved by the institutional review board or central ethics committee at each participating institution and was conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent. Patients were enrolled between 11 February 2013 and 6 July 2015 at 8 sites in Italy and Greece. Detailed information on the sites where patients were recruited is provided in supplemental Table 1 (available on the Blood Web site).
Patient demographics and baseline characteristics
| Initial luspatercept dose groups, mg/kg | Total (N = 64) | ||
|---|---|---|---|
| 0.2-0.4 (n = 12) | 0.6-1.25 (n = 52) | ||
| Age, median (range), y | 33.5 (22-53) | 41.0 (20-62) | 38.5 (20-62) |
| Female, n (%) | 5 (42) | 26 (50) | 31 (48) |
| Splenectomy, n (%) | 10 (83) | 33 (64) | 43 (67) |
| Non–transfusion-dependent patients, n | 12 | 21 | 33 |
| Hemoglobin, median (range), g/dL | 8.5 (7.0-9.6) | 8.5 (6.5-9.8) | 8.5 (6.5-9.8) |
| LIC, mean (SD), mg/g dry weight | 5.1 (2.9) | 5.6 (4.3) | 5.4 (3.8) |
| Transfusion-dependent patients, n | 0 | 31 | 31 |
| RBC transfusion burden, median (range), units/12 wk* | NA | 8.0 (4-18) | 8.0 (4-18) |
| LIC, mean (SD), mg/g dry weight† | NA | 5.0 (5.3) | 5.0 (5.3) |
| BMD | |||
| Total hip, mean (SD), z score | −1.1 (0.90) | −0.66 (1.27) | −0.74 (1.21) |
| Lumbar spine, mean (SD), z score | −1.62 (0.58) | −1.61 (0.95) | −1.61 (0.88) |
| FACIT-F score, median (range)‡ | NA | 41.5 (25-48) | 41.5 (25-48) |
| Initial luspatercept dose groups, mg/kg | Total (N = 64) | ||
|---|---|---|---|
| 0.2-0.4 (n = 12) | 0.6-1.25 (n = 52) | ||
| Age, median (range), y | 33.5 (22-53) | 41.0 (20-62) | 38.5 (20-62) |
| Female, n (%) | 5 (42) | 26 (50) | 31 (48) |
| Splenectomy, n (%) | 10 (83) | 33 (64) | 43 (67) |
| Non–transfusion-dependent patients, n | 12 | 21 | 33 |
| Hemoglobin, median (range), g/dL | 8.5 (7.0-9.6) | 8.5 (6.5-9.8) | 8.5 (6.5-9.8) |
| LIC, mean (SD), mg/g dry weight | 5.1 (2.9) | 5.6 (4.3) | 5.4 (3.8) |
| Transfusion-dependent patients, n | 0 | 31 | 31 |
| RBC transfusion burden, median (range), units/12 wk* | NA | 8.0 (4-18) | 8.0 (4-18) |
| LIC, mean (SD), mg/g dry weight† | NA | 5.0 (5.3) | 5.0 (5.3) |
| BMD | |||
| Total hip, mean (SD), z score | −1.1 (0.90) | −0.66 (1.27) | −0.74 (1.21) |
| Lumbar spine, mean (SD), z score | −1.62 (0.58) | −1.61 (0.95) | −1.61 (0.88) |
| FACIT-F score, median (range)‡ | NA | 41.5 (25-48) | 41.5 (25-48) |
NA, not applicable; SD, standard deviation.
Baseline RBC transfusion burden is defined as the total number of RBC units transfused in the 12 wk before first dose of luspatercept (and confirmed over 6 mo before treatment).
LIC data were not available at baseline for 1 of the 31 transfusion-dependent patients.
FACIT-F score was assessed in 26 of the 64 patients, none of whom received 0.2-0.4 mg/kg of luspatercept. For scoring assessment, a scale of 0 to 52 is used, with 0 indicating worst quality of life and 52, best. A score <44 points is considered to be a FACIT-F deficit.16
Patients
Patients with a documented diagnosis of β-thalassemia age ≥18 years were eligible. Those in the dose-escalation stage had either prior splenectomy or spleen size <18 cm at the longest diameter, as measured by abdominal ultrasound.
Exclusion criteria included folate deficiency, symptomatic splenomegaly, and history of thromboembolic events; individuals with adequate folate replacement therapy could be enrolled. Patients with any clinically significant pulmonary, cardiovascular, endocrine, neurologic, hepatic, gastrointestinal, infectious, immunologic, or genitourinary disease considered by the investigator to be inadequately controlled were also excluded.
Eligible patients were classified as either non–transfusion dependent or transfusion dependent. Patients not dependent on transfusions were eligible if they had a mean hemoglobin concentration of <10.0 g/dL (based on ≥2 measurements) and, if transfused, received <4 RBC units in the 8 weeks before study initiation. Patients with transfusion dependence were those who received an average of ≥4 RBC units every 8 weeks over the 6-month period before study initiation.
Study design
This phase 2, open-label, nonrandomized, uncontrolled, dose-finding study evaluated the effects of luspatercept in patients with β-thalassemia. Luspatercept was administered subcutaneously every 21 days at dose levels ranging from 0.2 to 1.25 mg/kg of body weight; this starting dose was selected based on preclinical and phase 1 clinical findings, including safety margin and pharmacodynamic markers.
The study was conducted in 2 stages: the initial stage and extension stage (Figure 1). In the dose-escalation cohorts of the initial stage, patients received up to 5 doses of luspatercept at 1 of 6 prespecified dose levels: 0.2, 0.4, 0.6, 0.8, 1.0, and 1.25 mg/kg. There was no formal sample size calculation used in the dose-finding stage. Up to 6 individuals were enrolled in each dose level, and dose cohorts were enrolled sequentially, from lowest to highest dose group. Once ≥3 patients in a dose cohort completed the study day-29 visit, response and AE data were reviewed by a safety review team comprising the coordinating investigator, medical monitor, and an independent hematologist. The safety review team made recommendations as to whether to enroll patients in the next highest dose cohort or proceed to the expansion cohort at a selected dose level.
![Figure 1. Study design and cohorts. The analytic sample size for the primary study end point was 63 patients (non–transfusion dependent [NTD], n = 31; transfusion dependent [TD], n = 32); this included all patients who received 0.6 to 1.25 mg/kg of luspatercept in either the initial or extension stage of the study. Doses <0.6 mg/kg of luspatercept were not considered efficacious. *Patients assigned to the dose-escalation cohorts received luspatercept at dose levels of 0.2 to 1.25 mg/kg; patients were assigned to the following dose levels: 0.2 mg/kg (n = 6 patients; all NTD), 0.4 mg/kg (n = 6 patients; all NTD), 0.6 mg/kg (n = 5 NTD patients; n = 1 TD patient), 0.8 mg/kg (n = 3 NTD patients; n = 3 TD patients), 1.0 mg/kg (n = 2 NTD patients; n = 4 TD patients), and 1.25 mg/kg (n = 1 NTD patient; n = 4 TD patients). †Patients assigned to the expansion cohort and the extension stage received luspatercept at dose levels of 0.8 to 1.25 mg/kg; patients were assigned to the expansion cohort after review of adverse event (AE) and efficacy data for patients in the dose-escalation cohorts. ‡Patients entering the extension with treatment interruption were those who had finished the initial stage of treatment and had completed the end-of-study visit at least 28 days before receiving the first dose in the extension. §Patients enter the 2-month follow-up after discontinuing from or completing the extension stage; because of the treatment period of 60 months, no patients have yet entered the follow-up period.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/12/10.1182_blood-2018-10-879247/4/m_blood879247f1.png?Expires=1763494472&Signature=2u3Bd-Z8o9BTDmZu4Wu5tEKnp6Y3d352EX6bmsdYajVTlDz7pZ3wt~-fApAG5bC4rIr~GphHaMdahAt8Qb~NA-~JHGO0VX2ZWITHBrUXWnCIOK3eazM9lP~qT7fQWNdanK6syFtHAw~~YaFX6nrm9z2LLw1fgTlPm2KpLWiHEj8a65htRSV124bsc34yFToYgi~-Tw9xVXA8DnNSWhseZoLRjhlNoBKId7sxpxPHs3IgRgUdxn~c2zWL~-F6N1hnXyW2pXKLY8M9kSXNU41d~KimFSEe6BaSk8DhtFXWRbB4RUZcX~s5tCXb1IWWtWBOmjlVa4gvo9ojPKd6YKcp5g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Study design and cohorts. The analytic sample size for the primary study end point was 63 patients (non–transfusion dependent [NTD], n = 31; transfusion dependent [TD], n = 32); this included all patients who received 0.6 to 1.25 mg/kg of luspatercept in either the initial or extension stage of the study. Doses <0.6 mg/kg of luspatercept were not considered efficacious. *Patients assigned to the dose-escalation cohorts received luspatercept at dose levels of 0.2 to 1.25 mg/kg; patients were assigned to the following dose levels: 0.2 mg/kg (n = 6 patients; all NTD), 0.4 mg/kg (n = 6 patients; all NTD), 0.6 mg/kg (n = 5 NTD patients; n = 1 TD patient), 0.8 mg/kg (n = 3 NTD patients; n = 3 TD patients), 1.0 mg/kg (n = 2 NTD patients; n = 4 TD patients), and 1.25 mg/kg (n = 1 NTD patient; n = 4 TD patients). †Patients assigned to the expansion cohort and the extension stage received luspatercept at dose levels of 0.8 to 1.25 mg/kg; patients were assigned to the expansion cohort after review of adverse event (AE) and efficacy data for patients in the dose-escalation cohorts. ‡Patients entering the extension with treatment interruption were those who had finished the initial stage of treatment and had completed the end-of-study visit at least 28 days before receiving the first dose in the extension. §Patients enter the 2-month follow-up after discontinuing from or completing the extension stage; because of the treatment period of 60 months, no patients have yet entered the follow-up period.
Study design and cohorts. The analytic sample size for the primary study end point was 63 patients (non–transfusion dependent [NTD], n = 31; transfusion dependent [TD], n = 32); this included all patients who received 0.6 to 1.25 mg/kg of luspatercept in either the initial or extension stage of the study. Doses <0.6 mg/kg of luspatercept were not considered efficacious. *Patients assigned to the dose-escalation cohorts received luspatercept at dose levels of 0.2 to 1.25 mg/kg; patients were assigned to the following dose levels: 0.2 mg/kg (n = 6 patients; all NTD), 0.4 mg/kg (n = 6 patients; all NTD), 0.6 mg/kg (n = 5 NTD patients; n = 1 TD patient), 0.8 mg/kg (n = 3 NTD patients; n = 3 TD patients), 1.0 mg/kg (n = 2 NTD patients; n = 4 TD patients), and 1.25 mg/kg (n = 1 NTD patient; n = 4 TD patients). †Patients assigned to the expansion cohort and the extension stage received luspatercept at dose levels of 0.8 to 1.25 mg/kg; patients were assigned to the expansion cohort after review of adverse event (AE) and efficacy data for patients in the dose-escalation cohorts. ‡Patients entering the extension with treatment interruption were those who had finished the initial stage of treatment and had completed the end-of-study visit at least 28 days before receiving the first dose in the extension. §Patients enter the 2-month follow-up after discontinuing from or completing the extension stage; because of the treatment period of 60 months, no patients have yet entered the follow-up period.
After the dose-escalation cohorts, an expansion cohort was enrolled. On the basis of recommendations of the safety review team, individuals in the expansion cohort received luspatercept at a starting dose level of 0.8 mg/kg, with dose titration up to 1.25 mg/kg allowed after completion of 2 treatment cycles. A sample of 30 evaluable patients in the expansion cohort was calculated by a priori power analysis to provide ∼87% power (with a 1-sided significance level of .05) to differentiate an erythroid response rate of 30% from a minimal erythroid response rate of 10%. Patients were on study for ∼24 weeks in the initial stage, including a 4-week screening period, 12-week treatment period, and 8-week follow-up period.
Patients completing the initial stage could be enrolled in the long-term extension stage to receive luspatercept for up to 5 years. Those without treatment interruption could directly roll over to the extension stage at a starting dose equal to their last dose level in the initial stage. Those with treatment interruption were reassessed for eligibility and transfusion status before starting the extension and received a starting dose level of 0.8 mg/kg. In this manner, patients classified as non–transfusion dependent in the base study could be reclassified as transfusion dependent upon entering the extension study, or vice versa, subject to transfusion status evaluation. Dose titration up to 1.25 mg/kg was allowed in the extension stage. Patients discontinuing or completing the extension stage entered a 2-month follow-up period. Additional study design and statistical information can be found in the supplemental Methods.
Study end points
Primary end point
The primary end point was erythroid response, defined as a hemoglobin increase from baseline of ≥1.5 g/dL for ≥2 weeks (in the absence of RBC transfusions) for non–transfusion-dependent patients and as a reduction in RBC transfusion burden over a 12-week interval of ≥20% as compared with pretreatment for transfusion-dependent patients.
Secondary end points
Secondary end points reported in this manuscript include hemoglobin increase of ≥1.5 g/dL from baseline for ≥12 consecutive weeks for non–transfusion-dependent patients, reduction in RBC transfusion burden of ≥33% or ≥50% for transfusion-dependent patients, time to and duration of erythroid response, and changes in liver iron concentration (LIC) measured using magnetic resonance imaging. The time to response was calculated from the day patients received the first dose to the beginning of the first 12-week rolling period in which they satisfied the response criterion.
The safety and tolerability of luspatercept was evaluated throughout the study, including AE profiling, clinical laboratory tests, and vital signs. Additional secondary end point assessments beyond the scope of this manuscript include the proportion of patients requiring no RBC transfusions for ≥8 consecutive weeks, additional iron metabolism parameters, erythropoiesis parameters, bone metabolism parameters, and hemolysis parameters.
Exploratory end points
Exploratory end points included quality of life, which was measured using the Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) patient-reported outcome tool. The FACIT-F scale ranges from 0 to 52, with 52 being the best quality of life.15 Other secondary end points reported included improvements in leg ulcers and bone mineral density (BMD) by dual-energy X-ray absorptiometry scan. Leg ulcer severity was scored using a scale of 0 to 5, where 0 denoted fully healed and 5 denoted gangrene, as well as with measurements; however, some leg ulcers were only graded by physical examination and measurements.16 Information on when and how the end points in this study were collected, including those not reported in this manuscript, can be found in supplemental Table 2.
Statistical methods
The primary efficacy end point of erythroid response is summarized by percentage of responders and its 95% exact confidence interval (CI). For secondary and exploratory efficacy end points, the continuous measurement data, including LIC, hemoglobin, duration of erythroid response, and BMD, are summarized by mean change from baseline and corresponding standard deviation (SD) and 95% CI, as required. The dichotomized response measurement data, including ≥33% and ≥50% RBC transfusion reduction from baseline, are summarized by percentage of response and 95% CI. Correlation between improvement in FACIT-F and hemoglobin change is summarized using the Pearson correlation coefficient and tested to determine the significance of this correlation. P values presented are based on a 2-sided test; P values <.05 were considered statistically significant. SAS statistical software was used (version 9.4; SAS Institute, Cary, NC). Changes from baseline hemoglobin values were further evaluated by mixed-effects analysis with baseline hemoglobin, investigational sites, and time; of these, sites and time were treated as random effects.
With regard to missing values when calculating the primary end point, a patient without adequate data for evaluation was considered a nonresponder. For example, any patient without 12-week RBC transfusion data after treatment was considered a nonresponder for the purpose of calculating erythroid response. For other patient data, including LIC, BMD, and quality of life, only data actually collected were used in the analysis; no imputation was performed.
Results
Patient characteristics
Sixty-four patients were enrolled between 11 February 2013 and 6 July 2015. In the initial stage, patients received luspatercept every 3 weeks for up to 5 doses (median exposure, 105 days; range, 21-107 days). Thirty-five of the 64 patients were enrolled in the dose-escalation cohorts (0.2-1.25 mg/kg), and 29 were enrolled in the expansion cohort (0.8-1.25 mg/kg). At baseline, 33 patients were non–transfusion dependent and 31 were transfusion dependent.
In the initial stage of the study, 12 patients received luspatercept at lower dose levels of 0.2 to 0.4 mg/kg and 52 patients at higher dose levels of 0.6 to 1.25 mg/kg; all 12 lower-dose patients were non–transfusion dependent. A total of 51 patients carried over to the long-term extension stage of the study, during which all patients received higher dose levels of 0.6 to 1.25 mg/kg; 25 of these patients had treatment interruption between treatment stages. Median total duration of treatment of all 64 patients was 428 days (range, 21-768 days); treatment of 33 patients was ongoing as of the 2 September 2016 data cutoff date. Of the 18 patients who discontinued the extension stage, 4 did so because of a medical reason or AE, 1 was lost to follow-up, 10 discontinued at patient request, 1 died as a result of a cerebrovascular accident not related to treatment, and 2 discontinued because of protocol noncompliance (Figure 1).
Of the 51 patients who continued to the extension stage, 11 had received lower dose levels in the initial stage. Thus, across both stages of the study, 63 of 64 patients received higher dose levels of 0.6 to 1.25 mg/kg of luspatercept at some point during the study; 31 were non–transfusion dependent, and 32 were transfusion dependent.
Patient demographics and baseline characteristics are listed in Table 1.16 Median age at baseline was 38.5 years (range, 20-62 years), and 48% of patients were women. Among patients who were non–transfusion dependent, median hemoglobin level at baseline was 8.5 g/dL (range, 6.5-9.8 g/dL), whereas mean (± SD) LIC was 5.4 ± 3.8 mg/g dry weight. Among patients with transfusion dependence, median baseline transfusion burden was 8.0 RBC units per 12 weeks (range, 4-18 units), and mean LIC was 5.0 ± 5.3 mg/g. In total, 43 patients (67%) had previously had a splenectomy.
Efficacy
Hemoglobin response in non–transfusion-dependent patients
Primary end point
Rates of erythroid response for non–transfusion-dependent patients are listed in Table 2. Of 31 non–transfusion-dependent patients treated with luspatercept at dose levels of 0.6 to 1.25 mg/kg, 18 (58%) achieved a mean hemoglobin increase from baseline of ≥1.5 g/dL over 14 consecutive days (95% CI, 39.1-75.5).
Response rates based on erythroid response criteria in patients with β-thalassemia
| 0.6-1.25 mg/kg luspatercept (N = 63)* | 95% CI | |
|---|---|---|
| Non–transfusion-dependent patients, n/N (%) | n = 31† | |
| Mean hemoglobin increase, g/dL | ||
| ≥1.5 for 14 d‡ | 18/31 (58) | 39.1-75.5 |
| ≥1.0 for 12 wk | 22/31 (71) | 52.0-85.8 |
| ≥1.5 for 12 wk | 14/31 (45) | 27.3-64.0 |
| Decrease in LIC ≥2 mg/g dry weight§ | 5/15 (33) | 11.8-61.6 |
| Transfusion-dependent patients, n/N (%) | n = 32† | |
| Transfusion burden reduction, % | ||
| ≥20‡ | 26/32 (81) | 63.6-92.8 |
| ≥33 | 23/32 (72) | 53.3-86.3 |
| ≥50 | 20/32 (63) | 43.7-78.9 |
| Decrease in LIC ≥2 mg/g dry weight§ | 5/9 (56) | 21.2-86.3 |
| 0.6-1.25 mg/kg luspatercept (N = 63)* | 95% CI | |
|---|---|---|
| Non–transfusion-dependent patients, n/N (%) | n = 31† | |
| Mean hemoglobin increase, g/dL | ||
| ≥1.5 for 14 d‡ | 18/31 (58) | 39.1-75.5 |
| ≥1.0 for 12 wk | 22/31 (71) | 52.0-85.8 |
| ≥1.5 for 12 wk | 14/31 (45) | 27.3-64.0 |
| Decrease in LIC ≥2 mg/g dry weight§ | 5/15 (33) | 11.8-61.6 |
| Transfusion-dependent patients, n/N (%) | n = 32† | |
| Transfusion burden reduction, % | ||
| ≥20‡ | 26/32 (81) | 63.6-92.8 |
| ≥33 | 23/32 (72) | 53.3-86.3 |
| ≥50 | 20/32 (63) | 43.7-78.9 |
| Decrease in LIC ≥2 mg/g dry weight§ | 5/9 (56) | 21.2-86.3 |
Results shown are the numbers of patients achieving any of the specified end points over any unbroken 12-wk period on study compared with baseline; only patients receiving dose levels of 0.6-1.25 mg/kg of luspatercept at any time during the study are shown (63 of 64 total patients; initial and extension stages).
One of the 64 patients initially enrolled is not included because the patient did not receive luspatercept ≥0.6 mg/kg during the study.
One patient classified as non–transfusion dependent during the initial stage of the study was reclassified as transfusion dependent before entering the extension stage and is counted as transfusion dependent in the table.
Primary end point; other end points shown are secondary end points.
Includes patients with baseline LIC ≥3 mg/g dry weight who were treated for ≥4 mo.
Secondary end points
Of 31 non–transfusion-dependent patients treated with luspatercept at dose levels of 0.6 to 1.25 mg/kg, 22 (71%) achieved a mean hemoglobin increase from baseline of ≥1.0 g/dL over 12 weeks (95% CI, 52.0-85.8) compared with the 12 weeks before treatment; 14 (45%) non–transfusion-dependent patients achieved a mean hemoglobin increase of ≥1.5 g/dL over 12 weeks (95% CI, 27.3-64.0). Of 10 non–transfusion-dependent patients with prior splenectomy receiving luspatercept at dose levels of 0.6 to 1.25 mg/kg, 8 (80%) achieved a mean hemoglobin increase from baseline of ≥1.5 g/dL over 12 weeks, compared with 6 (29%) of 21 patients without prior splenectomy. The relationship between splenectomy and response will be investigated further in a future trial.
Mean hemoglobin changes from baseline for those treated with luspatercept at dose levels of 0.2 to 0.4 mg/kg and 0.6 to 1.25 mg/kg in the initial stage of the study are shown in Figure 2A. Median time to hemoglobin response among non–transfusion-dependent patients in the initial stage of the study was 8 days (range, 7-30 days). Mean change in hemoglobin over the study duration, including the extension stage up to 18 months total duration, for all non–transfusion-dependent patients treated with dose levels of 0.6 to 1.25 mg/kg (n = 31) is shown in Figure 2B.

Mean (± 95% CI) change in hemoglobin level relative to baseline. (A) Non–transfusion-dependent patients with β-thalassemia treated with luspatercept at 0.2 to 0.4 mg/kg compared with 0.6 to 1.25 mg/kg during the initial stage of the study. (B) All non–transfusion-dependent patients with β-thalassemia treated with luspatercept at 0.6 to 1.25 mg/kg during the initial and/or extension stage of the study.
Mean (± 95% CI) change in hemoglobin level relative to baseline. (A) Non–transfusion-dependent patients with β-thalassemia treated with luspatercept at 0.2 to 0.4 mg/kg compared with 0.6 to 1.25 mg/kg during the initial stage of the study. (B) All non–transfusion-dependent patients with β-thalassemia treated with luspatercept at 0.6 to 1.25 mg/kg during the initial and/or extension stage of the study.
Transfusion burden reduction in transfused patients
Primary end point
Thirty-two patients with transfusion dependence were treated with luspatercept at dose levels of 0.6 to 1.25 mg/kg during the study. Of these, 26 (81%) achieved a transfusion burden reduction of ≥20% over any 12 weeks on study compared with the 12 weeks before baseline (95% CI, 63.6-92.8); these data are presented in Table 2.
Secondary end points
RBC transfusion burden reduction of ≥33% was achieved in 23 patients (72%; 95% CI, 53.3-86.3), and ≥50% reduction was achieved in 20 (63%; 95% CI, 43.7-78.9). Of 21 transfusion-dependent patients with prior splenectomy, 14 (67%) achieved a transfusion burden reduction of ≥20% over a fixed 12-week period vs baseline, compared with 4 (36%) of 11 patients without prior splenectomy.
Transfusion burden reduction relative to pretreatment of all transfused patients who had a baseline transfusion burden of ≥2 RBC units and received 0.6 to 1.25 mg/kg of luspatercept is shown in Figure 3. For transfused patients, mean transfusion burden (± SD) over the best recorded 12-week responses on treatment was 3.0 units (±3.76), compared with 7.6 units (±3.56) at baseline. The pretransfusion hemoglobin levels did not vary substantially on treatment as compared with pretreatment and ranged between 9 and 10 g/dL.
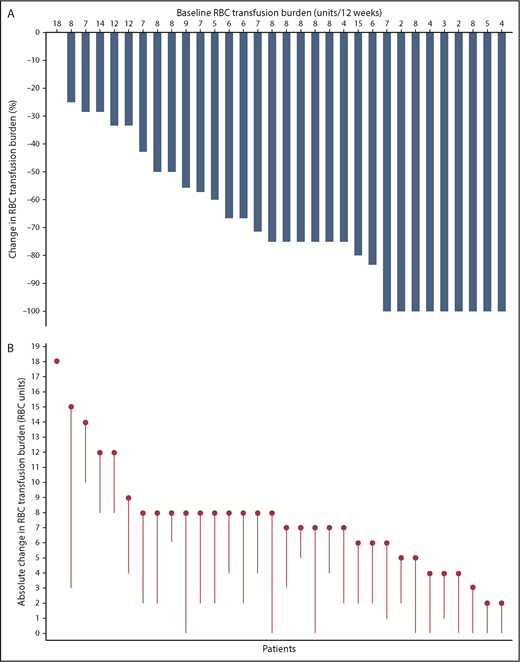
Transfusion burden reduction vs baseline for patients with β-thalassemia treated with luspatercept (n = 32). (A) Percentage change in RBC transfusion burden over a continuous 12-week period post baseline vs the 12-week baseline period. Each bar represents 1 patient; the best recorded continuous 12-week period of transfusion burden reduction post baseline was used to calculate the percentage change for each patient. (B) Absolute change in RBC units on study vs baseline. Each circle represents 1 patient’s baseline RBC transfusion burden, as absolute number of transfused units in a 12-week period; each line represents the best recorded transfusion burden reduction for the patient over a continuous 12-week period post baseline. For both panels, only patients with a baseline transfusion burden of ≥2 RBC units and 12-week postbaseline transfusion data are shown.
Transfusion burden reduction vs baseline for patients with β-thalassemia treated with luspatercept (n = 32). (A) Percentage change in RBC transfusion burden over a continuous 12-week period post baseline vs the 12-week baseline period. Each bar represents 1 patient; the best recorded continuous 12-week period of transfusion burden reduction post baseline was used to calculate the percentage change for each patient. (B) Absolute change in RBC units on study vs baseline. Each circle represents 1 patient’s baseline RBC transfusion burden, as absolute number of transfused units in a 12-week period; each line represents the best recorded transfusion burden reduction for the patient over a continuous 12-week period post baseline. For both panels, only patients with a baseline transfusion burden of ≥2 RBC units and 12-week postbaseline transfusion data are shown.
LIC
Secondary end point
Of 15 patients with non–transfusion-dependent β-thalassemia with baseline LIC ≥3 mg/g dry weight who were treated for ≥4 months, 5 (33%) achieved a decrease in LIC ≥2 mg/g dry weight (95% CI, 11.8-61.6; Figure 4A). Mean LIC (± SD) for non–transfusion-dependent patients at the end of the initial stage of treatment was −0.36 mg/g dry weight (±1.59), compared with 5.38 mg/g (±3.79) at baseline. Of 9 patients with transfusion dependence with baseline LIC ≥3 mg/g dry weight who were treated for ≥4 months, 5 (56%) achieved a decrease in LIC ≥2 mg/g dry weight (95% CI, 21.2-86.3; Figure 4B). Mean LIC (± SD) for transfusion-dependent patients at the end of the initial stage of treatment was −0.27 mg/g dry weight (±1.64), compared with 5.03 mg/g (±5.32) at baseline. All LIC responders were receiving ongoing concomitant iron chelation therapy.
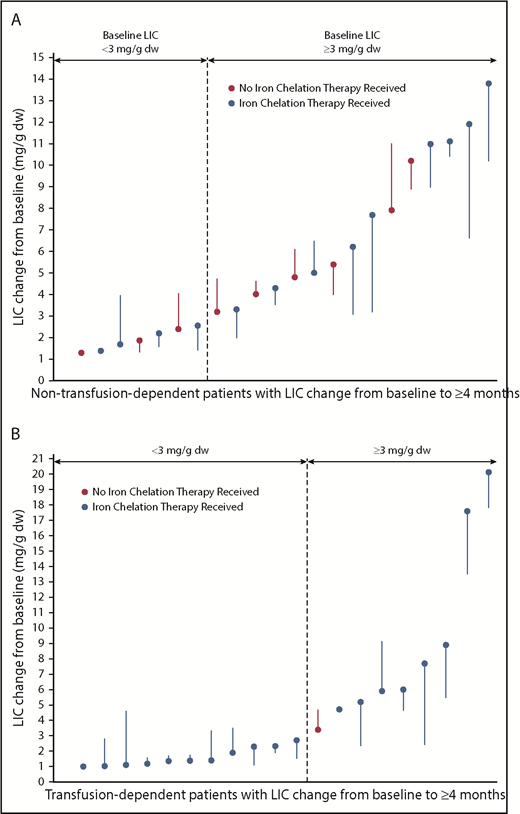
LIC change in non–transfusion-dependent (A) and transfusion-dependent β-thalassemia patients (B). Each filled circle represents 1 patient’s baseline LIC; each line represents the best recorded change in LIC post baseline. Only patients with ≥4 months postbaseline LIC are included. Patients noted to have received iron chelation therapy could have received it in the 84 days before treatment as well as on study. dw, dry weight.
LIC change in non–transfusion-dependent (A) and transfusion-dependent β-thalassemia patients (B). Each filled circle represents 1 patient’s baseline LIC; each line represents the best recorded change in LIC post baseline. Only patients with ≥4 months postbaseline LIC are included. Patients noted to have received iron chelation therapy could have received it in the 84 days before treatment as well as on study. dw, dry weight.
Exploratory end points
Leg ulcers
Six patients had from 1 to 6 leg ulcers over the malleoli at baseline. Each of the 6 patients had complete healing of at least 1 ulcer, with a total of 9 ulcers fully healed, 2 partially healed, and 3 not improved. Median time to complete healing was 10 weeks (range, 6-33 weeks). An example of ulcer healing is illustrated in Figure 5.
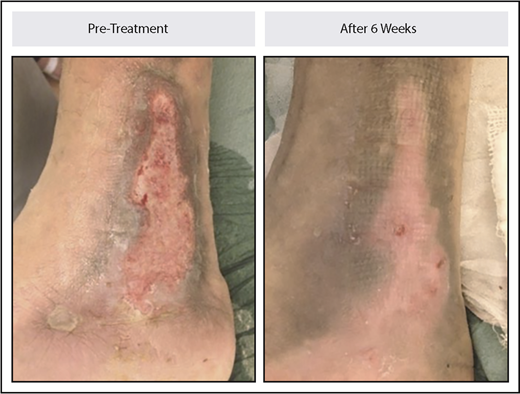
Leg ulcer healing in a patient with non–transfusion-dependent β-thalassemia treated with luspatercept. The patient received 0.4 mg/kg of luspatercept in the initial stage of the study and up to 1.25 mg/kg of luspatercept in the extension stage. The image example shown is representative of an ulcer that healed within 6 weeks.
Leg ulcer healing in a patient with non–transfusion-dependent β-thalassemia treated with luspatercept. The patient received 0.4 mg/kg of luspatercept in the initial stage of the study and up to 1.25 mg/kg of luspatercept in the extension stage. The image example shown is representative of an ulcer that healed within 6 weeks.
BMD
Those receiving luspatercept were assessed for changes in BMD. On baseline dual-energy X-ray absorptiometry scans, mean (± SD) BMD z score was −0.74 ± 1.21 for total hip (n = 25) and −1.61 ± 0.88 for lumbar spine (n = 27). After 24 weeks of treatment at dose levels ≥0.6 mg/kg, mean percentage change in BMD was +5.3% ± 15.8% for total hip (n = 14; P = .23) and +3.1% ± 6.6% for lumbar spine (n = 17; P = .07).
Quality of life
Quality-of-life changes were measured using the FACIT-F patient-reported outcome tool.15 Improvements in FACIT-F score in non–transfusion-dependent patients were significantly correlated with mean 12-week change in hemoglobin (n = 21; r = 0.64; P = .002). Of the non–transfusion-dependent patients with a baseline FACIT-F deficit (<44 points) compared with normative data,17 7 (78%) of 9 improved by ≥3 points at 24 weeks. A majority of these patients (86%) also had an improvement in mean hemoglobin of ≥1.0 g/dL over a 12-week period. As expected, we did not observe any significant changes in transfusion-dependent patients.
AEs
AEs assessed as treatment related are listed in Table 3. Grade 1 to 2 AEs were common, mainly during the first 8 weeks, with a clear trend to decrease. The most frequent were bone pain, headache, myalgia, arthralgia, musculoskeletal pain, back pain, and injection site pain. Grade 3 AEs considered treatment related were uncommon, with 6 reported in 5 patients (8%): 3 reports of grade 3 bone pain, 2 of grade 3 asthenia, and 1 of grade 3 headache. No treatment-related grade 4 AEs or serious AEs were reported. One patient with a history of cardiac disease died as a result of cardiac arrest considered unrelated to treatment; no treatment-related deaths were reported. Four (6%) of 64 patients discontinued treatment because of AEs. AEs were more frequent during the first 8 weeks, with a trend to decrease over time. Dose-delay and full AE information can be found in the supplemental Results and supplemental Table 3, respectively. Results pertaining to luspatercept pharmacokinetics are located in the supplemental Results as well.
AEs considered related to treatment occurring during the study (N = 64)
| AE preferred term* | Related TEAE, n (%) | |
|---|---|---|
| Grade 1-2 in ≥2 patients | Grade 3† | |
| Bone pain | 21 (33) | 3 (5) |
| Headache | 16 (25) | 1 (2) |
| Myalgia | 13 (20) | 0 |
| Arthralgia | 12 (19) | 0 |
| Musculoskeletal pain | 10 (16) | 0 |
| Back pain | 7 (11) | 0 |
| Injection site pain | 7 (11) | 0 |
| Asthenia | 6 (9) | 2 (3) |
| Erythroblastosis | 3 (5) | 0 |
| Injection site erythema | 3 (5) | 0 |
| Musculoskeletal chest pain | 3 (5) | 0 |
| Neck pain | 3 (5) | 0 |
| Pain in jaw | 3 (5) | 0 |
| Pyrexia | 3 (5) | 0 |
| Chills | 2 (3) | 0 |
| Influenza | 2 (3) | 0 |
| Injection site pruritus | 2 (3) | 0 |
| Macules | 2 (3) | 0 |
| Muscle spasms | 2 (3) | 0 |
| Nausea | 2 (3) | 0 |
| Pain in extremity | 2 (3) | 0 |
| Paresthesia | 2 (3) | 0 |
| AE preferred term* | Related TEAE, n (%) | |
|---|---|---|
| Grade 1-2 in ≥2 patients | Grade 3† | |
| Bone pain | 21 (33) | 3 (5) |
| Headache | 16 (25) | 1 (2) |
| Myalgia | 13 (20) | 0 |
| Arthralgia | 12 (19) | 0 |
| Musculoskeletal pain | 10 (16) | 0 |
| Back pain | 7 (11) | 0 |
| Injection site pain | 7 (11) | 0 |
| Asthenia | 6 (9) | 2 (3) |
| Erythroblastosis | 3 (5) | 0 |
| Injection site erythema | 3 (5) | 0 |
| Musculoskeletal chest pain | 3 (5) | 0 |
| Neck pain | 3 (5) | 0 |
| Pain in jaw | 3 (5) | 0 |
| Pyrexia | 3 (5) | 0 |
| Chills | 2 (3) | 0 |
| Influenza | 2 (3) | 0 |
| Injection site pruritus | 2 (3) | 0 |
| Macules | 2 (3) | 0 |
| Muscle spasms | 2 (3) | 0 |
| Nausea | 2 (3) | 0 |
| Pain in extremity | 2 (3) | 0 |
| Paresthesia | 2 (3) | 0 |
Maximum severity reported per patient per preferred term.
TEAE, treatment-emergent AE.
Classified according to the Medical Dictionary for Regulatory Activities. Results shown are the numbers of patients reporting the specified TEAEs.
No grade 4 AEs considered related to luspatercept treatment were reported.
Discussion
In this phase 2 study of patients with β-thalassemia, the primary finding was the achievement of protocol-defined erythroid response, which occurred in 71% of non–transfusion-dependent patients and 81% of transfusion-dependent patients receiving higher doses of luspatercept. Population pharmacokinetic modeling suggests that luspatercept at 1.0 mg/kg is an appropriate starting dose for additional studies in patients with β-thalassemia, with titration up to 1.25 mg/kg if required to achieve optimal exposure.18
Luspatercept is a novel recombinant fusion protein containing a modified activin receptor type IIB (ActRIIB, ACVR2B) that acts as a ligand trap to block inhibitors of late-stage erythropoiesis.12 In this manner, luspatercept functions as an erythroid maturation agent to restore RBC production and ameliorate anemia. To date, most AEs reported in patients with β-thalassemia receiving luspatercept have been grade 1 or 2; few patients reported grade 3 to 4 AEs. In this study, the most frequent treatment-related AE, bone pain, was more commonly observed in patients with transfusion dependence and may have been due in part to the withdrawal of transfusions in response to treatment. Collection of longer-term AE data is ongoing in the extension stage of the study.
In this study, most patients with low FACIT-F quality-of-life fatigue scores at baseline had improved scores after luspatercept treatment. Most patients with low baseline fatigue scores who achieved an erythroid response had improvements in FACIT-F score during the study. Although the FACIT-F questionnaire focuses on the symptoms of anemia, transfusion-dependent patients were also assessed, and no significant change in score was observed posttreatment. This could be expected for patients who were asymptomatic at baseline with hemoglobin levels maintained during treatment.
Iron overload affects patients with β-thalassemia regardless of whether they are transfusion dependent or non–transfusion dependent. In non–transfusion-dependent β-thalassemia patients, abnormal regulation of intestinal absorption leads to iron overload, whereas in patients with transfusion dependence, iron overload occurs mostly as a result of frequent RBC transfusions.3,5 Despite the availability of iron chelation therapies, mean baseline LIC determined by magnetic resonance imaging was found to be abnormal in both non–transfusion-dependent and transfusion-dependent patients, with a mean concentration of 5.4 and 5.0 mg/g dry weight, respectively. Luspatercept treatment was associated with clinically significant decreases in LIC, particularly in patients who had baseline LIC ≥3 mg/g dry weight and who were receiving concomitant iron chelation therapy; the relative contributions of luspatercept and iron chelation therapy cannot be resolved.
Erythroid expansion resulting from ineffective erythropoiesis in β-thalassemia can lead to skeletal deformities and decreased BMD,19 which improved with luspatercept treatment in mouse models of the disease.14 Patients in this study had low mean baseline total hip and lumbar spine BMD z scores, with increases during treatment with luspatercept that were not found to be statistically significant.
Severe anemia, ineffective erythropoiesis, iron overload, and hypercoagulable state are potential risk factors for developing leg ulcers in β-thalassemia, particularly in nontransfused patients.20 Of the 6 patients with leg ulcers, which were often long-standing and resistant to other therapies, all had complete healing of 1 or more leg ulcers, which may have been due to drug effects on anemia, ineffective erythropoiesis, and/or iron overload, as well as transforming growth factor β superfamily signaling pathways; the mechanism is the subject of further ongoing research.
The study has several limitations. First, it had a single-group, open-label design without a control group for comparison. However, the lower-dose groups were considered subtherapeutic. Second, the study had a relatively small number of patients treated at different dose levels. Higher-dose groups with similar responses were pooled for this reason. Third, many patients had normal baseline BMD; the small numbers of patients with low BMD limited the analysis and interpretation of the effects of luspatercept. Fourth, the notable decreases in LIC were confounded by patients receiving concomitant chronic treatment with iron chelation therapy. Nonetheless, decreases in LIC may have been due in part to the decrease of transfusion burden and/or increase in iron utilization related to luspatercept treatment.
In conclusion, in this open-label, nonrandomized, uncontrolled dose-finding study of patients with β-thalassemia, a high percentage had improvement in hemoglobin or transfusion burden after use of luspatercept. These findings support a randomized clinical trial to assess efficacy and safety.
Data requests may be submitted to Celgene at www.celgeneclinicaldatasharing.com and must include a description of the research proposal.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ellis Neufeld, the independent hematologist on the safety review team, from Boston Children’s Hospital, who received compensation from Acceleron Pharma. The authors also wish to thank personnel at Acceleron Pharma, who helped manage the trial, including clinical operations, program management, statistical programming, data management, and adverse event monitoring: Chris Rovaldi, Ashley Leneus, Brett O’Hare, Tad Akers, and Dawn Wilson, who was an employee of Acceleron Pharma at the time of study. The authors also thank personnel at Chiltern International Ltd, funded by Acceleron Pharma: Carlo Lanza (executive medical officer/medical monitor) and Fabienne Durand Van der Schueren (global project manager).
This work was supported by Acceleron Pharma, and Celgene Corporation provided funding for this study. The authors received editorial and writing assistance from Daniel Gilmartin from Excerpta Medica, supported by Acceleron Pharma.
Acceleron Pharma was involved at every stage of the study, participating in the design and conduct of the study (including development of the study protocol and statistical analysis plan); collection, management, analysis, and interpretation of the data; and preparation, review, and approval of the manuscript. The decision to submit the manuscript for publication was made by all authors.
Authorship
Contribution: A. Piga had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; A. Piga, K.M.A., A.L., and M.L.S. were responsible for study conception and design; all authors were responsible for acquisition, analysis, or interpretation of data; A. Piga, K.M.A., A.F., M.L.S., I.T., and E.V. drafted the manuscript; all authors were responsible for critical revision of the manuscript for important intellectual content; A.L. and X.Z. performed statistical analysis; M.L.S. obtained funding; C.B.-P. provided administrative, technical, or material support; A. Piga, K.M.A., and M.L.S. supervised the study; and K.M.A. acted as medical monitor.
Conflict-of-interest disclosure: A. Piga has received research funding from Acceleron Pharma and Celgene Corporation; S.P. has received speaker’s honoraria from Novartis Oncology, has received research funding from Novartis Oncology and Acceleron Pharma, and has served on advisory boards at Bluebird Bio; E.V. has received research funding from Acceleron Pharma and Celgene Corporation; I.T. has received speaker’s honoraria from Novartis Oncology and has served on advisory boards at Bluebird Bio; C.B.-P. has received research funding from Acceleron Pharma and Celgene Corporation; X.Z., M.L.S., and K.M.A. are employed by and have equity ownership in Acceleron Pharma; and A.L. is employed by and has equity ownership in Celgene Corporation. The remaining authors declare no competing financial interests.
Correspondence: Antonio Piga, Department of Clinical and Biological Sciences, Turin University, Regione Gonzole, 10, 10043 Orbassano, Turin, Italy; e-mail: antonio.piga@unito.it.
