Key Points
PAPD5 inhibition stabilizes TERC, rescues telomerase, and lengthens telomeres in X-linked DC hESCs.
Modulation of PAPD5 improves definitive hematopoietic development from hESCs with a pathological mutation in dyskerin.

Abstract
Reduced levels of TERC, the telomerase RNA component, cause dyskeratosis congenita (DC) in patients harboring mutations in TERC, PARN, NOP10, NHP2, NAF1, or DKC1. Inhibition of the noncanonical poly(A) polymerase PAPD5, or the exosome RNA degradation complex, partially restores TERC levels in immortalized DKC1 mutant cells, but it remains unknown if modulation of posttranscriptional processing of TERC could improve hematopoietic output in DC. We used human embryonic stem cells (hESCs) with a common dyskerin mutation (DKC1_A353V), which have defective telomere maintenance and reduced definitive hematopoietic potential, to understand the effects of reducing EXOSC3 activity, or silencing PAPD5-mediated oligoadenylation, on hematopoietic progenitor specification and function in DC. Reduction of EXOSC3 or PAPD5 levels in DKC1 mutant hESCs led to functional improvements in TERC levels and telomerase activity, with concomitant telomere elongation and reduced levels of DNA damage signaling. Interestingly, the silencing of PAPD5, but not EXOSC3, significantly restored definitive hematopoietic potential in DKC1 mutant cells. Mechanistically, we show that PAPD5 inhibition is sustained in differentiated CD34+ cells, with a concomitant increase in mature, functional, forms of TERC, indicating that regulation of PAPD5 is a potential strategy to reverse hematologic dysfunction in DC patients.
Introduction
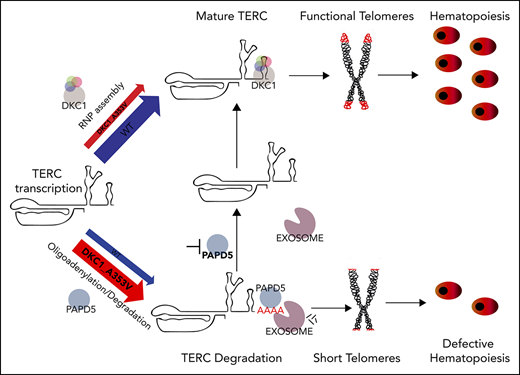
Patients with dyskeratosis congenita (DC) harbor mutations in telomere maintenance genes1,2 and suffer morbidity from bone marrow failure.3 Several of the mutations found in DC cause reduced TERC levels, resulting in telomerase impairment.4-9 Although overexpression of TERC increases hematopoietic output from DC cells,10 it is not a viable approach for patients. The discovery that TERC degradation by the exosome complex can be controlled by its oliogoadenylation status, through modulation of PAPD5 (noncanonical poly(A) polymerase 5), opened a new avenue of opportunity for clinical intervention in DC.11-14 However, it remains unknown if the reduction of TERC decay by modulation of PAPD5 or the exosome11-15 could restore hematopoietic potential in DC, a crucial end point in this disease.
We used human embryonic stem cells (hESCs) to assess the effect of silencing PAPD5 or the Exosome Component gene 3 (EXOSC3) on primitive and definitive hematopoietic potential of DC. We used hESCs harboring a common DKC1_A353V mutation, which recapitulates key aspects of the hematopoietic defects of DC.10 We show that silencing of PAPD5 or EXOSC3 increases telomerase activity, elongates telomeres, and reduces γH2AX in DKC1_A353V hESCs. However, only the silencing of PAPD5 and not EXOSC3 restored definitive hematopoietic potential in DKC1 mutants. Our data give strong support for the development of therapeutics targeting the posttranscriptional regulation of TERC by PAPD5 in patients with mutations that impair TERC stability.
Study design
Results and discussion
We and others have established that the hematopoietic differentiation of DKC1_A353V hESCs recapitulates major phenotypes of DC.10,18 To determine if TERC levels could be posttranscriptionally regulated in hESCs with clinically relevant mutations in DKC1, we treated genetically engineered DKC1_A353 hESCs10 with small interfering RNAs against EXOSC3 or PAPD5. Transient silencing of EXOSC3 or PAPD5 increased TERC levels (supplemental Figure 1A-B, available on the Blood Web site), prompting us to constitutively silence these genes. We targeted the AAVS1 safe-harbor locus19 of both wild-type (WT) and DKC1_A353V hESCs with short hairpin RNAs (shRNAs) against PAPD5 and EXOSC3 (Figure 1A), creating WT_shPAPD5, WT_shEXOSC3, DKC1_A353V_shPAPD5, and DKC1_A353V_shEXOSC3 hESCs. These cells showed significantly reduced levels of EXOSC3 and PAPD5 messenger RNAs (Figure 1B; supplemental Figure 1C) and protein (Figure 1C).

Modulation of EXOSC3 and PAPD5 rescue telomere integrity in DKC1_A353V hESCs. (A) Schematic depicting shRNA cassette insertion into the AAVS1 locus of hESCs. shRNA sequences used for each cassette are described in supplemental Methods and supplemental Table 1. HA, homology arm; RES, resistance cassette. (B) Quantification of EXOSC3 (left) and PAPD5 (right) levels in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 by quantitative reverse transcription polymerase chain reaction. (C) Western blot for EXOSC3 and PAPD5 in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs. LE, long exposure. β-Actin is shown as loading control. Quantification of band intensities is shown (relative to β-actin). (D) Quantification of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 by quantitative reverse transcription polymerase chain reaction. (E) Relative abundance of oligoadenylated reads at mature 3′ end of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESC. (F) Telomerase activity by telomere repeat amplification protocol in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs. Range of concentrations represents fourfold serial dilutions. L.C., loading control. (G) Telomere length analysis by telomere restriction fragment (TRF) of WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs at different passages. Passage numbers are described for each lane. For shEXOSC3 and shPAPD5 transfected cells, passage numbers reflect passage at transfection (35), plus number of passages since transduction. Quantification of mean telomere length is shown. (H) Quantification of interphase quantitative fluorescence in situ hybridization analysis, cells at same passage number as panel G. At least 40 nuclei were analyzed in each cell line. (I) Representative immunoblot analysis of γH2AX in WT, DKC1_A353V (passage 57), DKC1_A353V_shEXOSC3 (passage 35+28), and DKC1_A353V_shPAPD5 (passage 35+28) hESCs. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as a loading control. Numbers indicate band intensity relative to GAPDH. All experiments were conducted using n = 3, mean ± standard error of the mean, *P ≤ .05, unless otherwise indicated. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s post hoc test or by Bonferroni posttest in panel H. In panel H, ***P ≤ .0001. n.s., not significant; ZFN, zinc finger nucleases.
Modulation of EXOSC3 and PAPD5 rescue telomere integrity in DKC1_A353V hESCs. (A) Schematic depicting shRNA cassette insertion into the AAVS1 locus of hESCs. shRNA sequences used for each cassette are described in supplemental Methods and supplemental Table 1. HA, homology arm; RES, resistance cassette. (B) Quantification of EXOSC3 (left) and PAPD5 (right) levels in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 by quantitative reverse transcription polymerase chain reaction. (C) Western blot for EXOSC3 and PAPD5 in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs. LE, long exposure. β-Actin is shown as loading control. Quantification of band intensities is shown (relative to β-actin). (D) Quantification of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 by quantitative reverse transcription polymerase chain reaction. (E) Relative abundance of oligoadenylated reads at mature 3′ end of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESC. (F) Telomerase activity by telomere repeat amplification protocol in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs. Range of concentrations represents fourfold serial dilutions. L.C., loading control. (G) Telomere length analysis by telomere restriction fragment (TRF) of WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESCs at different passages. Passage numbers are described for each lane. For shEXOSC3 and shPAPD5 transfected cells, passage numbers reflect passage at transfection (35), plus number of passages since transduction. Quantification of mean telomere length is shown. (H) Quantification of interphase quantitative fluorescence in situ hybridization analysis, cells at same passage number as panel G. At least 40 nuclei were analyzed in each cell line. (I) Representative immunoblot analysis of γH2AX in WT, DKC1_A353V (passage 57), DKC1_A353V_shEXOSC3 (passage 35+28), and DKC1_A353V_shPAPD5 (passage 35+28) hESCs. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as a loading control. Numbers indicate band intensity relative to GAPDH. All experiments were conducted using n = 3, mean ± standard error of the mean, *P ≤ .05, unless otherwise indicated. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s post hoc test or by Bonferroni posttest in panel H. In panel H, ***P ≤ .0001. n.s., not significant; ZFN, zinc finger nucleases.
TERC levels were significantly increased by constitutive silencing of PAPD5 or EXOSC3 in DKC1_A353V but not in WT hESCs (Figure 1D; supplemental Figure 1D). Targeted RNA sequencing at the 3′ end of TERC showed that DKC1_A353V_shPAPD5 cells have a significant reduction in the percentage of oligo(A) species at the mature (Figure 1E) and extended (supplemental Figure 2A) forms of TERC, when compared with WT, DKC1_A353V, and DKC1_A353V_shEXOSC3 hESCs. This demonstrates silencing of PAPD5 and EXOSC3 in hESCs rescues TERC levels by reducing its 3′ adenylation- and exosome-mediated degradation. The DKC1_A353V mutation by itself did not cause a change in the mature TERC composition in terms of oligo(A) reads (supplemental Figure 2B), supporting a model where any unassembled TERC is rapidly degraded.14
Modulation of 3′ oligoadenylation by PAPD5, as well as the inhibition of EXOSC3, also increased telomerase activity (Figure 1F; supplemental Figure 3A) and telomere length (Figure 1G-H; supplemental Figure 3B) in DKC1_A353V_shPAPD5 and DKC1_A353V_shEXOSC3 hESCs. Cells with silenced PAPD5 or EXOSC3 show reduced γH2AX (Figure 1I), indicating lower levels of DNA damage signaling, a common phenotype of DC.20 Thus, posttranscriptional modulation of TERC restores major defects of DKC1_A353V mutants.
We next examined if modulation of PAPD5 and EXOSC3 could restore the hematopoietic output of DKC1_A353V cells. Impaired definitive hematopoietic potential in DKC1_A353V cells can be rescued by overexpression of TERC.10 We hypothesized that the silencing of PAPD5 or EXOSC3 could also rescue definitive hematopoiesis in DKC_A353V hESCs. We performed serum-free differentiations to independently derive primitive and definitive hematopoietic progenitors by stage-specific modulation of WNT (supplemental Figure 4A).16,17 Silencing of PAPD5 and EXOSC3 does not affect early stages of primitive or definitive hematopoietic development in WT cells (supplemental Figure 4B-I). Likewise, colony-forming potential of both hematopoietic programs is normal (Figure 2A-B), indicating that a reduction in exosome-mediated RNA degradation is not detrimental to hematopoiesis in WT settings.
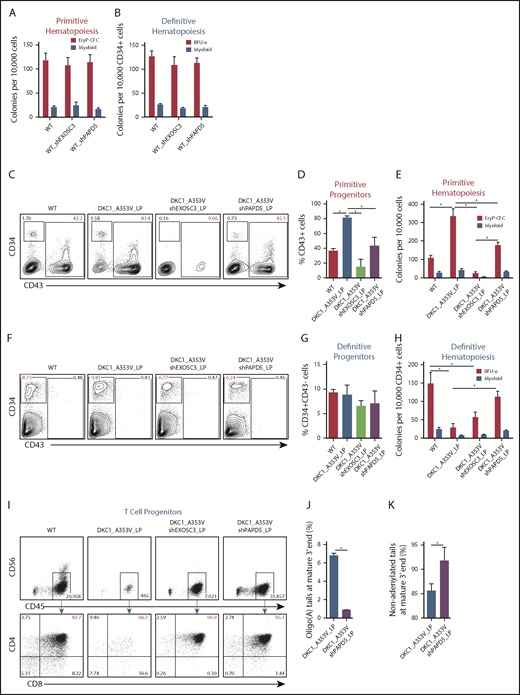
PAPD5 silencing restores defective hematopoiesis in DKC1_A353V cells. (A) Colony-forming cell (CFC) potential of primitive hematopoietic progenitors in WT, WT_shEXOSC3, and WT_shPAPD5 cells from day 11 of IWP2-derived specification. (B) CFC potential of definitive hematopoietic progenitors in WT, WT_shEXOSC3, and WT_shPAPD5 cells from day 8 sorted CD34+CD43− populations, as described in supplemental Figure 3A. (C) Representative flow cytometric analysis of CD34 and CD43 expression on day 11 of differentiation, following IWP2 treatment in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 cells. (D) Quantification of CD43+ population obtained from day 11 differentiation cultures treated with IWP2, as in panel C. (E) Primitive CFC potential in day 11 differentiation cultures, as in panel C. (F) Representative flow cytometric analysis of CD34 and CD43 expression on day 8 of definitive differentiation, following CHIR99021 and SB-431542 treatment in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 cells. (G) Quantification of CD34+CD43− population obtained from day 8 differentiation cultures treated with CHIR99021 and SB-431542, as in panel F. (H) CFC potential of definitive hematopoietic progenitors, generated as shown in supplemental Figure 3A. (I) T-cell potential of CD34+CD43− populations derived from WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5, obtained following CHIR99021 and SB-431542 treatment. (J) Relative abundance of oligoadenylated reads at mature 3′ end of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESC. n = 2. (K) Relative abundance of nonadenylated reads at mature 3′ end of TERC as in panel J. All experiments were conducted using n = 3, mean ± standard error of the mean, *P ≤ .05, unless otherwise indicated. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s post hoc test or Student t test (J-K). In red, population of interest. In all panels, LP denotes late passage (passage number >55).
PAPD5 silencing restores defective hematopoiesis in DKC1_A353V cells. (A) Colony-forming cell (CFC) potential of primitive hematopoietic progenitors in WT, WT_shEXOSC3, and WT_shPAPD5 cells from day 11 of IWP2-derived specification. (B) CFC potential of definitive hematopoietic progenitors in WT, WT_shEXOSC3, and WT_shPAPD5 cells from day 8 sorted CD34+CD43− populations, as described in supplemental Figure 3A. (C) Representative flow cytometric analysis of CD34 and CD43 expression on day 11 of differentiation, following IWP2 treatment in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 cells. (D) Quantification of CD43+ population obtained from day 11 differentiation cultures treated with IWP2, as in panel C. (E) Primitive CFC potential in day 11 differentiation cultures, as in panel C. (F) Representative flow cytometric analysis of CD34 and CD43 expression on day 8 of definitive differentiation, following CHIR99021 and SB-431542 treatment in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 cells. (G) Quantification of CD34+CD43− population obtained from day 8 differentiation cultures treated with CHIR99021 and SB-431542, as in panel F. (H) CFC potential of definitive hematopoietic progenitors, generated as shown in supplemental Figure 3A. (I) T-cell potential of CD34+CD43− populations derived from WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5, obtained following CHIR99021 and SB-431542 treatment. (J) Relative abundance of oligoadenylated reads at mature 3′ end of TERC in WT, DKC1_A353V, DKC1_A353V_shEXOSC3, and DKC1_A353V_shPAPD5 hESC. n = 2. (K) Relative abundance of nonadenylated reads at mature 3′ end of TERC as in panel J. All experiments were conducted using n = 3, mean ± standard error of the mean, *P ≤ .05, unless otherwise indicated. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s post hoc test or Student t test (J-K). In red, population of interest. In all panels, LP denotes late passage (passage number >55).
We next examined the consequences of EXOSC3 and PAPD5 silencing specifically during the primitive hematopoietic specification of DKC1_A353V hESCs. Analysis of mesoderm (KDR+CD235a+) on day 3 of differentiation showed that all hESC lines behaved similarly at this stage (supplemental Figure 5A-B). However, confirming our previous data,10 at day 11 (Figure 2C-D; CD43+ cells), as well as at the terminal primitive myeloid and erythroid colony potential assessment (Figure 2E), DKC1_A353V cells displayed increased differentiation capacity relative to WT and DKC1_A353V_shPAPD5 cells. This increased primitive hematopoietic potential of DKC1_A353V hESCs, which we hypothesize is a reflection of stress erythropoiesis,21 is also reduced when TERC is overexpressed,10 indicating that modulation of PAPD5 mimics the functional consequences of TERC overexpression during primitive differentiation of DKC1 mutants. On the other hand, unlike WT cells (Figure 2A-B), silencing of EXOSC3 is detrimental during primitive hematopoiesis of DKC1_A353V hESCs, because these fail to specify into primitive CD43+ progenitors (Figure 2C-D), leading to minimal erythroid and myeloid potential (Figure 2E). We hypothesize the toxicity observed in DKC1_A353V_shEXOSC3 cells is related to the essential role of the exosome in processing and destruction of different RNA classes,22 which could deter its clinical use in DC.
As bone marrow failure in DC is caused by defective definitive hematopoietic specification, we analyzed the consequences of PAPD5 and EXOSC3 silencing in DKC1_A353V cells during that developmental program. Although day 3 mesoderm (supplemental Figure 5C-D; KDR+CD235-− cells) and day 8 CD34+CD43− cells (Figure 2F-G) were similar in all samples, definitive colony potential analysis showed compromised colony-forming potential in DKC1_A353V cells (Figure 2H). However, silencing of PAPD5 (but not EXOSC3) significantly increased the hematopoietic potential in DKC1_A353V_shPAPD5 cells, to levels similar to WT (Figure 2H). Globin expression patterns confirm these populations were derived from definitive, and not primitive, hematopoiesis (supplemental Figure 5E). In addition, although DKC1_A353V cells had a compromised ability to give rise to CD4+CD8+ T-cell progenitors, DKC1_A353V_shPAPD5 cells displayed a clear increase in CD4+CD8+ cellularity (Figure 2I). These observations provide compelling evidence that silencing PAPD5 increases definitive, multilineage, hematopoietic potential in DKC1_A353V mutants. Finally, consistent with PAPD5 rescuing differentiation by affecting the oligoadenylation of TERC, 3′-end sequencing from day 8 definitive CD34+CD43− populations shows that PAPD5 silencing leads to a reduction in oligo(A) species in mature TERC (Figure 2J; supplemental Figure 6), with a concomitant increase in the total number of nonadenylated TERC reads (Figure 2K) in CD34+ cells.
Our data provide molecular and functional evidence that modulation of PAPD5 restores in vitro hematopoiesis in DKC1_A353V mutants, through direct regulation of the 3′-end maturation of TERC. Likely, a similar strategy could be employed to rescue hematopoiesis in cells with different mutations in DKC1, or harboring mutations in other genes that also lead to reduced levels of mature TERC, a hypothesis that should be further tested experimentally. In addition, although our data have not indicated any toxicity associated with the silencing of PAPD5 during hematopoiesis in WT or DKC1 mutants, future studies aiming at the identification of potential targets of PAPD5 in the hematopoietic system, as well as their implication for blood development, should be performed. As current therapeutic alternatives for bone marrow failure in DC remain largely ineffective, the posttranscriptional regulation of TERC by PAPD5 might represent a novel avenue for the management of this disease.
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by a National Institutes of Health, National Heart, Lung, and Blood Institute T32 training grant in molecular hematology (HL007088 [W.C.F.]), the Philip Majerus Fellowship Fund (A.T.V.), the National Science Foundation (K.A.B.), Howard Hughes Medical Institute (R.P. and S.S.), National Institutes of Health, National Institute of General Medical Sciences grant R01GM45443 (R.P. and S.S.), an American Society of Hematology Scholar Award (C.M.S.), National Institutes of Health, National Heart, Lung, and Blood Institute grants 4R00HL114732 and 1R01HL137793 (L.F.Z.B.), and grants from the V Foundation for Cancer Research (L.F.Z.B.), the Edward Mallinckrodt Jr Foundation (L.F.Z.B.), the AA&MDS International Foundation (L.F.Z.B.), the CONCERN Foundation (L.F.Z.B.), the American Federation for Aging Research (L.F.Z.B.), the Longer Life Foundation (L.F.Z.B.), the Center for Regenerative Medicine at Washington University in St. Louis (L.F.Z.B.), and a grant from the Department of Defense Bone Marrow Failure Research Program (BM160054 [L.F.Z.B. and C.M.S.]).
Authorship
Contribution: W.C.F., S.S., A.T.V., R.P., C.M.S., and L.F.Z.B. designed the experiments and analyzed the data; W.C.F., S.S., A.T.V., and K.A.B. performed the experiments; and W.C.F., S.S., R.P., C.M.S., and L.F.Z.B. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luis Francisco Zirnberger Batista, Hematology Division, Washington University in St. Louis, Campus Box 8125, Washington University School of Medicine, 660 South Euclid Ave, St. Louis, MO 63110; e-mail: lbatista@wustl.edu.