Key Points
−21M individuals harbor better-educated NKG2A+ NK cells with superior degranulation capacity.
HLA-B dimorphism was associated with improved outcome in −21M patients during IL-2–based immunotherapy.

Abstract
Natural killer (NK) cell function is regulated by inhibitory receptors, such as the family of killer immunoglobulin-like receptors (KIRs) and the NKG2A/CD94 heterodimer. These receptors recognize cognate HLA class I molecules on potential target cells, and recent studies imply that an HLA-B dimorphism at position −21 in the gene segment encoding the leader peptide dictates whether NK cell regulation primarily relies on the KIRs or the NKG2A/CD94 receptor. The impact of this HLA-B dimorphism on NK cell–mediated destruction of leukemic cells or on the course of leukemia is largely unknown. In a first part of this study, we compared functions of NK cells in subjects carrying HLA-B −21M or 21T using interleukin-2 (IL-2)–activated NK cells and leukemic cells from patients with acute myeloid leukemia (AML). Subjects carrying HLA-B −21M harbored better-educated NKG2A+ NK cells and displayed superior capacity to degranulate lytic granules against KIR ligand-matched primary leukemic blasts. Second, we aimed to define the potential impact of HLA-B −21 variation on the course of AML in a phase 4 trial in which patients received IL-2–based immunotherapy. In keeping with the hypothesis that 21M may be associated with improved NK cell functionality, we observed superior leukemia-free survival and overall survival in −21M patients than in −21T patients during IL-2–based immunotherapy. We propose that genetic variation at HLA-B −21 may determine the antileukemic efficacy of activated NK cells and the clinical benefit of NK cell–activating immunotherapy.
Introduction
Acute myeloid leukemia (AML) is characterized by the rapid expansion of immature myeloid cells in the bone marrow and peripheral blood. Despite achieving complete remission (CR) after repeated courses of chemotherapy, the majority of patients experience relapse of leukemia with poor prospects of long-term survival. Eligible high-risk patients may receive allogeneic stem cell transplants (allo-SCTs),1 but there is no consensually efficacious treatment available to prevent relapse in nontransplanted patients.2 The benefit of allo-SCT in AML points to the capability of the immune system to eliminate leukemic cells, and several studies have highlighted a role for natural killer (NK) cells in AML.3-5
NK cells are innate cytotoxic cells that kill malignant cells without prior sensitization.6 NK cells spare healthy cells that express appropriate levels of HLA class I molecules that engage with germline-encoded inhibitory NK cell receptors, such as killer cell immunoglobulin-like receptors (KIRs) and the CD94/NKG2A (NKG2A) heterodimeric receptor. The genes encoding KIRs and their HLA ligands are highly polymorphic, and polymorphisms in HLA class I genes have created 3 major epitopes that are recognized by KIRs. The HLA-C alleles are defined by a dimorphism that defines every HLA-C allele as C1 or C2, recognized by KIR2DL2/L3 and KIR2DL1, respectively. The third epitope, Bw4, is encoded by a subset of HLA-A and HLA-B alleles and is recognized by KIR3DL1. Depending on inheritance and differences between alleles, it is possible for each individual to harbor genes for 1, 2, or 3 epitopes that can be recognized by KIRs.7,8 Notably, HLA alleles display differential expression, and the KIRs bind their ligands with varying affinity. Thus, the signaling strength from the different KIR–HLA pairs will differ.
In addition to KIRs, NK cells are regulated by NKG2A, which recognizes the nonclassical HLA class I molecule HLA-E. The folding of HLA-E requires binding of a peptide derived from the leader sequence of HLA class I; thus, HLA-E expression is a sensor for the cellular production of HLA-ABC.9,10
Inhibitory receptors have a seemingly paradoxical role in NK cell biology; on the one hand, they inhibit NK cell effector responses against target cells, and, on the other hand, they set the functional competence of an individual NK cell. Thus, an NK cell that exists in an HLA environment where it receives strong input from inhibitory receptors will react more vigorously to a cell lacking inhibitory ligands compared with an NK cell that is receiving less inhibitory input at steady-state. This process, through which NK cells gain function by inhibitory signals, is known as licensing or education.11-13 However, several reports have indicated that immune perturbations by cytokines, therapeutic antibodies, and other therapies can make up for a less-educated state and, thus, allow poorly educated NK cell populations to exert effector functions.14-17 Accordingly, NK cells may override the inhibitory signals conveyed by the interaction between NKG2A and its low-expressed ligand HLA-E,18,19 suggesting that the thresholds for education and inhibition can be skewed by immune perturbations.
It was recently reported that NK cell education is predominantly controlled by NKG2A or KIRs, depending on a dimorphism in the HLA-B gene.20 In contrast to HLA-A and HLA-C alleles, which consistently encode a leader peptide presentable on HLA-E, only a few alleles of HLA-B generate peptides that bind to HLA-E. This is due to a dimorphism at position −21 in the leader sequence; the peptide containing a methionine (−21M) can bind to HLA-E, whereas the threonine variant (−21T) does not.21 Accordingly, individuals with HLA-B −21M alleles have higher surface expression of HLA-E.20 Interestingly, haplotypes carrying HLA-B −21M rarely contain a Bw4 domain or have a gene encoding HLA-C2; instead, they encode certain low-expressed HLA-C1 variants.20 Collectively, these data indicate that NK cell function is predominantly regulated either by KIRs or by NKG2A in a given individual. Apart from studies in HIV,22-24 little is known about the clinical consequences of this dichotomy.
In the present study, we sought to determine to what extent NK cell responses to leukemic cells are differentially regulated by NKG2A and KIRs in the presence or absence of cytokine stimulation. We further aimed to investigate whether the HLA-B −21 dimorphism, which dictates dependence on NKG2A or KIRs, affects the capacity to target leukemic cells and impacts the clinical outcome of cytokine-based immunotherapy. We show that physiological levels of HLA-E were relatively poor at blocking degranulation responses by interleukin-2 (IL-2)–stimulated NK cells. Furthermore, individuals carrying the genotype encoding HLA-B −21M/x harbored more educated NKG2A+ NK cells, and these cells displayed a superior capacity to degranulate against KIR ligand–matched leukemic blasts. In line with these results, AML patients carrying HLA-B −21M/x displayed superior leukemia-free survival (LFS) and overall survival (OS) after IL-2–based immunotherapy. Collectively, our study sheds light on the role of the interplay among licensing, cytokine-induced activation, and inhibitory receptor signaling for NK cell control of leukemic cells.
Methods
Isolation of human leukocytes and cell lines
Buffy coats from healthy donors were obtained from the blood center at Sahlgrenska University Hospital. Peripheral blood mononuclear cells (PBMCs) were purified using dextran sedimentation, followed by density gradient centrifugation (Lymphoprep; STEMCELL Technologies). PBMCs were further separated into NK cells using a MACS NK Cell Isolation Kit, according to the manufacturer’s instructions (Miltenyi Biotec).
AML cells were obtained from newly diagnosed patients at the Sahlgrenska University Hospital, and PBMCs were purified as described above. Leukemic blasts were generally defined as CD34+ cells and sorted on a BD FACSAria using CD34-PE (clone 8G12; BD Biosciences).
K562 cells were maintained in Iscoves’ medium with 10% fetal calf serum and 1% l-glutamine. 221-WT and 221-AEH cells (kindly provided by Karl-Johan Malmberg, Oslo University Hospital) were maintained in RPMI 1640 with 10% fetal calf serum and 1% l-glutamine, as well as with 1% Hygromycin B Gold (InvivoGen) for AEH cells. HLA-E–transfected T2 cells (T2/HLA-E cells; kindly provided by Christina Bade-Döding, Hannover Medical School) were maintained as described previously.25
Degranulation and cytokine production assays
In degranulation assays, prestimulated NK cells (IL-2; 100-1000 U/mL; Chiron; for 1-3 days) were incubated with target cells (K562, 221-WT, 221-AEH, T2 cells or AML blasts) in the presence of anti-CD107a. In T2 cell assays, the HLA-A leader peptide (VMAPRTLVL) was used to maintain HLA-E expression.25 After 4 hours, cells were stained with combinations of the following antibodies: CD3–APC–H7 (SK7), CD56-BV786 (3G8), CD107a-BUV395 (H4A3), granzyme B–Bv421 (GB11), interferon-γ (IFN-γ)–BUV395 (B27), tumor necrosis factor α (TNF-α)–Alexa Fluor-488 (Mab11; all from BD Biosciences), NKG2A-PE (Z199), KIR2DL1/S1–PE-Cy7 (EB6), KIR2DL2/L3–PE-Cy5.5 (GL183; all from Beckman Coulter), HLA-E–PE-Cy7 (3D12), KIR3DL1-APC (DX9; both from BioLegend), and LIVE/DEAD Violet (Life Technologies). In experiments analyzing intracellular cytokines, BD GolgiPlug was added after 1 hour. Analyses were carried out using a BD LSRFortessa SORP instrument.
DNA extraction, KIR/HLA ligand, and HLA-B/C genotyping
DNA was purified from whole blood using a Roche MagNA Pure 96 System and from PBMCs using a QIAGEN DNeasy Blood & Tissue Kit. KIR ligands were determined using an Olerup SSP KIR HLA Ligand kit. The HLA-B and -C allele genotype was determined using the respective LABType SSO Class I Locus Typing Test from One Lambda. Data were collected using a Bio-Plex 200 system from Bio-Rad (Luminex IS 2.3 software) and analyzed using HLA Fusion 4.1 software. NK cells and AML cells were defined as KIR ligand matched based on ligand presence or absence of HLA-C1/C2/Bw4. Designation of M or T HLA-B allele was determined from HLA-B allele number and information obtained from Horowitz et al.20
Patients
Eighty-four patients with AML in first CR (18-79 years) were enrolled in a single-armed multicenter phase 4 study (Re:Mission, NCT01347996). Patients received 10 consecutive 21-day cycles of histamine dihydrochloride (HDC; 0.5 mg, subcutaneously twice daily) and low-dose IL-2 (16 400 IU/kg, subcutaneously twice daily) for 18 months or until relapse/death. Primary end points for the study included assessment of NK cell and T cell dynamics before and after treatment cycles. Results for primary end points have been published previously,17,26-29 and detailed patient characteristics can be found elsewhere.28 Results presented in this article were obtained post hoc. The trial was approved by ethics committees from each participating institution and was conducted according to the principles of the Declaration of Helsinki. All patients gave written informed consent before enrollment in the study. Samples collected before treatment and after the first treatment cycle are referred to as cycle 1 day 1 and cycle 1 day 21, respectively.
Statistical analyses
All statistical analyses for results from the Re:Mission study were performed according to the statistical trial plan. Unpaired, 2-sided Student t tests were used to compare NK cell phenotypes between genotype groups. One-way analysis of variance, followed by the Bonferroni multiple-comparison test, was used for multiple comparisons. Statistical analysis of OS and LFS was performed using the log-rank test. Parameters that significantly predicted LFS and/or OS were further analyzed by univariable and multivariable Cox regression analysis. Univariable analyses were performed for classical prognosis markers, such as age, risk group classification, number of induction courses required to achieve CR (1 or >1), number of consolidation courses (0-2 or >2), and previously reported NK cell–related biomarkers (NKp46 expression, cytomegalovirus serostatus, KIR/HLA discordance).17,28,30 Covariates with values of P < .1 (age, induction courses, cytomegalovirus, NKp46) were included in the multivariable analysis.
Results
NKG2A expression is elevated in AML patients in remission and is further induced during immunotherapy
Therapeutic blockade of inhibitory T-cell receptors has proven a successful strategy in various types of malignancies. As mentioned above, NK cells are equipped with similar ITIM-coupled receptors (eg, KIRs and NKG2A), and the expression of these receptors in NK cells may be of relevance for the NK cell–mediated eradication of malignant cells. For the cohort of AML patients in first CR included in the Re:Mission trial (NCT01347996), we performed extensive NK cell phenotyping, with emphasis on the expression of inhibitory receptors. NK cells from AML patients and healthy donors displayed pronounced differences with regard to expression patterns of inhibitory receptors; foremost, the percentage of NKG2A+ NK cells was strikingly elevated in AML patients (Figure 1A). This was not merely reflecting a more immature NK cell population in AML patients, because NKG2A expression was not restricted to KIR− or single-KIR+ NK cell subsets. Instead, NK cells expressing multiple KIRs also displayed elevated proportions of NKG2A+ cells. The frequencies of NKG2A+ subsets increased further during HDC/IL-2 immunotherapy (Figure 1B). To investigate whether IL-2 was responsible for the induced NKG2A expression, we performed in vitro experiments in which healthy donor NK cells were exposed to IL-2 (500 U/mL) for 3 days. The frequency of NKG2A+ cells was significantly increased on NK cell subsets expressing 0, 1, or 2 KIRs upon IL-2 stimulation (Figure 1C). In line with this finding, an NK cell transcriptome analysis (NCBI Gene Expression Omnibus database, accession number GSE8059 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE8059)31 demonstrated elevated levels of NKG2A transcripts in NK cells after 2, 8, and 24 hours of IL-2 stimulation (Figure 1D). Interestingly, transcript levels of other inhibitory receptors, such as PD-1, CTLA-4, Tim-3, and Lag-3, displayed similar patterns, whereas transcripts encoding the KIR family of receptors decreased in response to IL-2 stimulation (supplemental Figure 1, available on the Blood Web site). Along with the induction of NKG2A, IL-2 was found to induce expression of the cytotoxic mediator granzyme B, especially in NKG2A+ subsets of NK cells (Figure 1E-F; supplemental Figure 1).
![Figure 1. NKG2A and granzyme B distribution and responses of NK cells in vivo and in vitro. (A) Distribution of NK cell subsets in AML patients before treatment start (n = 64) and in healthy donors (n = 24). Frequency of NKG2A+ NK cells in subsets expressing 0, 1, 2, or 3 KIRs in AML patients (n = 54) before or after cycle 1 (B) and in unstimulated and IL-2–stimulated (500 U/ml) healthy donors (n = 6) (C). (D) Fold change, compared with start, in messenger RNA expression of NKG2A after IL-2 stimulation. (E) Granzyme B expression (median fluorescence intensity [MFI]) in unstimulated and IL-2–stimulated NK cell subsets (n = 24). (F) Percentage increase in granzyme B response between unstimulated and IL-2–stimulated NK cells (n = 24). (G) LFS for patients dichotomized by median NKG2A expression at treatment start (low, n = 29 or high, n = 30). (H) LFS for patients dichotomized by median frequency of NKG2A+ NK cells (low, n = 29 or high, n = 30). *P < .05, **P < .005, ***P < .0005.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/13/10.1182_blood-2018-09-874990/3/m_blood874990f1.png?Expires=1768845409&Signature=MhfQWMn7J7BUBVwhCMTQcsS-LXZiMHiMXtpQAJMY4oDwPXXIN8eJIV64sPl94Y0zkIBk0t0gnIadr-B~i0ImH8Eiv9umQN0ywbJBgo3bLGBpS6zWELhwwL4or1QV3Seb6nuyg3KNqcZ96xWAkBmAiPLuCjg3HPPOQU5u75rIg8WO~tY-EI6zJ995gataVKdj2v5IneMpk9ycbfhZ-PNjiozb5JovkL5P9W23u9m11xfgM4g5o7tJme35Up5WADmK6wAUxVGu8m3AB8oJUXoIoxCO5Cs9C3mF23oVPlJmJfdL~UbxwzbRVhRgzktHGw2ycu5IYVkwi6FGA~1DkB53ww__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
NKG2A and granzyme B distribution and responses of NK cells in vivo and in vitro. (A) Distribution of NK cell subsets in AML patients before treatment start (n = 64) and in healthy donors (n = 24). Frequency of NKG2A+ NK cells in subsets expressing 0, 1, 2, or 3 KIRs in AML patients (n = 54) before or after cycle 1 (B) and in unstimulated and IL-2–stimulated (500 U/ml) healthy donors (n = 6) (C). (D) Fold change, compared with start, in messenger RNA expression of NKG2A after IL-2 stimulation. (E) Granzyme B expression (median fluorescence intensity [MFI]) in unstimulated and IL-2–stimulated NK cell subsets (n = 24). (F) Percentage increase in granzyme B response between unstimulated and IL-2–stimulated NK cells (n = 24). (G) LFS for patients dichotomized by median NKG2A expression at treatment start (low, n = 29 or high, n = 30). (H) LFS for patients dichotomized by median frequency of NKG2A+ NK cells (low, n = 29 or high, n = 30). *P < .05, **P < .005, ***P < .0005.
NKG2A and granzyme B distribution and responses of NK cells in vivo and in vitro. (A) Distribution of NK cell subsets in AML patients before treatment start (n = 64) and in healthy donors (n = 24). Frequency of NKG2A+ NK cells in subsets expressing 0, 1, 2, or 3 KIRs in AML patients (n = 54) before or after cycle 1 (B) and in unstimulated and IL-2–stimulated (500 U/ml) healthy donors (n = 6) (C). (D) Fold change, compared with start, in messenger RNA expression of NKG2A after IL-2 stimulation. (E) Granzyme B expression (median fluorescence intensity [MFI]) in unstimulated and IL-2–stimulated NK cell subsets (n = 24). (F) Percentage increase in granzyme B response between unstimulated and IL-2–stimulated NK cells (n = 24). (G) LFS for patients dichotomized by median NKG2A expression at treatment start (low, n = 29 or high, n = 30). (H) LFS for patients dichotomized by median frequency of NKG2A+ NK cells (low, n = 29 or high, n = 30). *P < .05, **P < .005, ***P < .0005.
Recent studies demonstrated that AML patients with low expression of HLA-ABC in the myeloid compartment show superior outcome after HDC/IL-2 immunotherapy27 and that a lack of inhibitory KIR ligands was associated with a favorable outcome.17 Collectively, these data suggest that a reduced inhibitory input to NK cells is associated with improved survival. The observed induction of NKG2A expression should be associated with increased inhibitory input, and we hypothesized that AML patients with high levels of NKG2A+ cells would have a higher relapse risk and inferior survival. However, as shown in Figure 1G, no difference was observed between patients with high- or low-intensity expression of NKG2A among NKG2A+ cells. By contrast, there was a trend toward improved survival among patients with an above-median percentage of NKG2A+ cells (Figure 1H).
HLA-E inhibition is aberrant in leukemic cells and threshold dependent
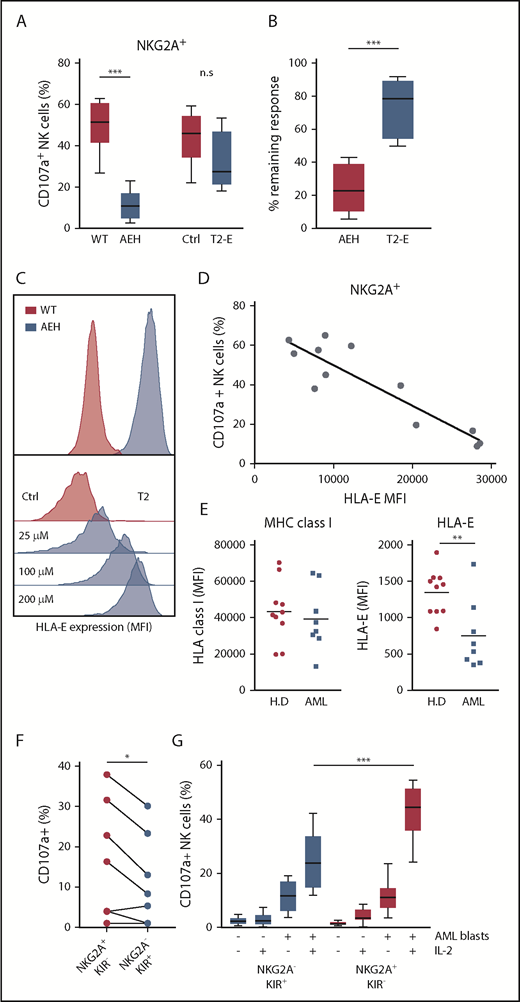
The above results led us to investigate the role of NKG2A–HLA-E interactions in NK cells’ capacity to kill leukemic cells. Earlier studies have demonstrated the impact of HLA-E inhibition on NKG2A+ NK cells using a 721.221 cell line transfected with a construct comprising the HLA-E molecule with a bound peptide from HLA-A (referred to as 221-AEH cells).21 In accordance with previous studies,32,33 221-AEH cells strongly inhibited degranulation responses in IL-2–activated NKG2A+ NK cells. However, when comparing HLA-E expression in 221-AEH cells and primary AML blasts, we observed that 221-AEH cells had a 10-fold higher expression of HLA-E. In further experiments, we took advantage of T2/HLA-E cells, in which HLA-E expression can be modulated by varying the concentration of exogenously added HLA-E–presentable peptides. Using different peptide concentrations, we observed an inverse correlation between degranulation by NKG2A+ NK cells and T2/HLA-E cell expression of HLA-E. Thus, a more physiologically relevant expression level of HLA-E on T2/HLA-E cells could still significantly inhibit degranulation responses in unstimulated NK cells (supplemental Figure 2). However, degranulation by IL-2–stimulated NK cells toward these cells was largely intact, suggesting that IL-2 prestimulation enables NK cells to withstand physiologically relevant HLA-E/NKG2A–mediated inhibitory signals (Figure 2A-D).
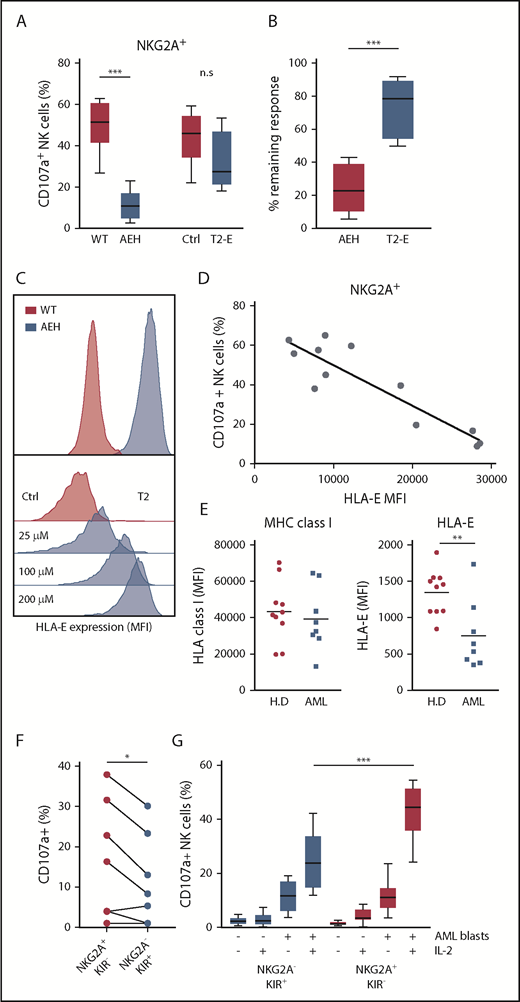
NKG2A+ NK cell responses toward HLA-E–expressing cell lines and AML blasts. (A) Degranulation responses of IL-2–stimulated NKG2A+ NK cells (n = 8) toward .221-WT, .221-AEH, T2 cells without HLA peptide (Ctrl), and T2 cells with HLA peptide (T2-E). (B) Comparison of remaining response of NKG2A+ NK cells with the presence of HLA-E compared with the response observed in the HLA-E− control cell lines. (C) HLA-E expression in .221-WT and .221-AEH cells and titration curve with HLA peptide of HLA-E expression in T2 cells. (D) Percentage of CD107a+ cells among IL-2–activated NKG2A+ NK cells after exposure to T2 cells with indicated HLA-E expression. (E) Expression of MHC class I and HLA-E on PBMCs from healthy donors (n = 10) and on CD34+ AML blasts (n = 8). (F) Frequency of CD107a+ NK cells in indicated NK cell subsets against HLA-matched CD34+ AML blasts from 8 patients. (G) Frequency of CD107a+ NK cells (unstimulated, n = 10; IL-2 stimulated, n = 12) in indicated NK cell subsets against HLA-matched CD34+ AML blasts from 2 patients that triggered substantial degranulation in (F). n.s, not significant. *P < .05, **P < .005, ***P < .0005.
NKG2A+ NK cell responses toward HLA-E–expressing cell lines and AML blasts. (A) Degranulation responses of IL-2–stimulated NKG2A+ NK cells (n = 8) toward .221-WT, .221-AEH, T2 cells without HLA peptide (Ctrl), and T2 cells with HLA peptide (T2-E). (B) Comparison of remaining response of NKG2A+ NK cells with the presence of HLA-E compared with the response observed in the HLA-E− control cell lines. (C) HLA-E expression in .221-WT and .221-AEH cells and titration curve with HLA peptide of HLA-E expression in T2 cells. (D) Percentage of CD107a+ cells among IL-2–activated NKG2A+ NK cells after exposure to T2 cells with indicated HLA-E expression. (E) Expression of MHC class I and HLA-E on PBMCs from healthy donors (n = 10) and on CD34+ AML blasts (n = 8). (F) Frequency of CD107a+ NK cells in indicated NK cell subsets against HLA-matched CD34+ AML blasts from 8 patients. (G) Frequency of CD107a+ NK cells (unstimulated, n = 10; IL-2 stimulated, n = 12) in indicated NK cell subsets against HLA-matched CD34+ AML blasts from 2 patients that triggered substantial degranulation in (F). n.s, not significant. *P < .05, **P < .005, ***P < .0005.
Previous studies show that HLA-A and HLA-B are ≥5 times more abundant than HLA-C (ligands to KIR), which, in turn, is 25 times more abundant than the NKG2A ligand HLA-E.34 AML blasts displayed variable expression levels of HLA-ABC but typically expressed comparable levels as healthy PBMCs, whereas HLA-E expression on AML blasts was found to be significantly lower (Figure 2E). Based on the difference in relative expression of the inhibitory ligands, we speculated that NK cells would more easily override NKG2A-dependent inhibition than KIR-mediated inhibition in assays against primary AML blasts. As shown in Figure 2F, after IL-2 stimulation, the NKG2A+KIR− subset showed significantly more degranulation in comparison with the NKG2A−KIR+ subset toward KIR ligand–matched AML blasts from 8 AML patients (Figure 2F). To allow a firm analysis of the contribution of the different NK cell subsets, we chose 2 patients (one C1/C1 and one C2/C2) whose blasts triggered substantial NK cell degranulation and performed degranulation experiments with NK cells from a series of KIR ligand–matched donors. Although unstimulated NK cells mounted only a modest degranulation response to KIR ligand–matched AML blasts, IL-2 stimulation allowed the NKG2A+KIR− subset to degranulate significantly more than the NKG2A−KIR+ subset (Figure 2G). Taken together, these data suggest that activated NK cells may more easily overcome NKG2A-mediated inhibition than KIR-mediated inhibition if the corresponding ligands are expressed at physiological levels.
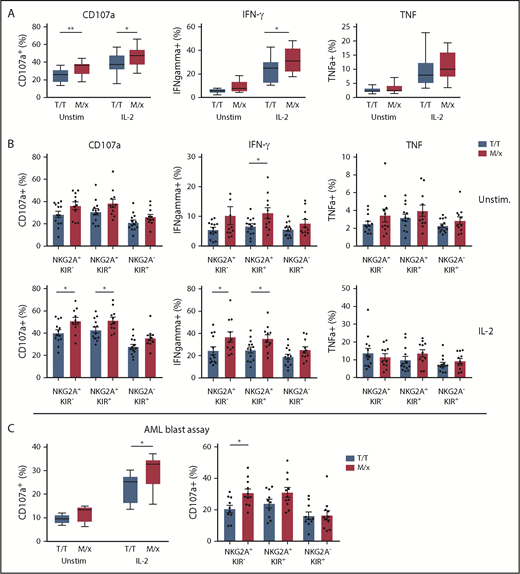
As mentioned above, NK cells in a given individual are predominantly regulated by NKG2A or KIRs, depending on a dimorphism in HLA-B.20 The presence of ≥1 copy of M in HLA-B alleles has been reported to shift the phenotype toward NKG2A-dependent inhibition, whereas T/T individuals have NK cells that are predominantly regulated by KIRs. Next, we set out to investigate how a predominant NK cell regulation by NKG2A or KIR translated into response against HLA-deficient targets and primary leukemic blasts. We genotyped 24 healthy donors for HLA-B and grouped donors into M/x and T/T genotypes. In a first series of experiments, purified NK cells from the genotyped donors were cocultured with HLA class I− K562 cells and analyzed for degranulation (CD107a) and cytokine production (IFN-γ and TNF-α). Overall, NK cells from M/x donors degranulated significantly more vigorously against K562 cells than did NK cells from T/T donors, when left unstimulated and after IL-2 prestimulation. A significantly higher proportion of NK cells from M/x donors produced IFN-γ after IL-2 stimulation (Figure 3A). When NK cells were gated based on maturation stages into 3 major groups (NKG2A+KIR−, NKG2A+KIR+, NKG2A−KIR+), only NKG2A+ subsets from M/x donors were significantly more functional than their counterparts from T/T donors (Figure 3B). These results imply that NK cells from M/x donors have better educated NKG2A+ NK cells, which impacts on the overall NK cell response toward HLA class I− target cells.
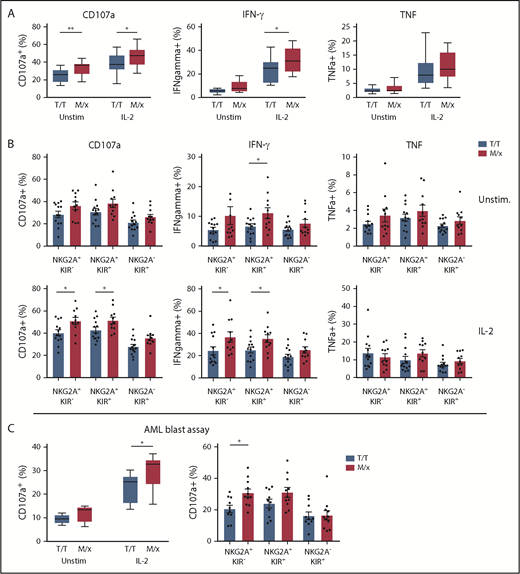
NK cell educational responses based on HLA-B −21. (A) Frequency of responding NK cells toward K562 cells in terms of CD107a, IFN-γ, and TNF-α positivity in unstimulated and IL-2–stimulated NK cells from donors with an M/x (red, n = 11) or T/T (white, n = 13) genotype. (B) Frequency of responding NK cell subsets in the K562 assay as in (A). (C) Frequency of CD107a+ NK cells against HLA-matched CD34+ AML blasts using unstimulated and IL-2–stimulated NK cells from M/x (red, n = 11) or T/T (white, n = 10) donors (left panel). Responses in IL-2–stimulated NK cell subsets (right panel). *P < .05, **P < .005.
NK cell educational responses based on HLA-B −21. (A) Frequency of responding NK cells toward K562 cells in terms of CD107a, IFN-γ, and TNF-α positivity in unstimulated and IL-2–stimulated NK cells from donors with an M/x (red, n = 11) or T/T (white, n = 13) genotype. (B) Frequency of responding NK cell subsets in the K562 assay as in (A). (C) Frequency of CD107a+ NK cells against HLA-matched CD34+ AML blasts using unstimulated and IL-2–stimulated NK cells from M/x (red, n = 11) or T/T (white, n = 10) donors (left panel). Responses in IL-2–stimulated NK cell subsets (right panel). *P < .05, **P < .005.
Next, we investigated whether the HLA-B −21 dimorphism influenced the capacity of NK cells to target KIR ligand–matched HLA class I–intact AML blasts. In accordance with the results shown in Figure 3C, unstimulated NK cells displayed little degranulation against AML blasts. However, when prestimulated with IL-2, NK cells obtained from M/x donors degranulated significantly more against AML blasts than did NK cells from T/T donors (Figure 3C). Breaking down the degranulation response into the 3 major subpopulations of CD56dim NK cells showed that NKG2A+ populations accounted for the enhanced degranulation among NK cells from M/x donors.
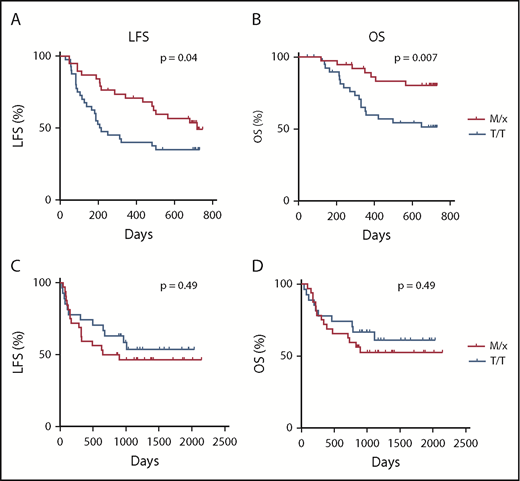
The superior degranulation responses of IL-2–stimulated NK cells from M/x donors against primary AML blasts motivated us to investigate whether clinical outcome in AML was affected by the HLA-B −21 genotype. Therefore, we determined the HLA-B genotype and the −21 dimorphism status in 80 AML patients enrolled in the Re:Mission phase 4 trial receiving HDC/IL-2. As shown in Figure 4A and B, patients carrying HLA-B −21 M/x genotype displayed superior LFS (P = .04, log-rank test; P = .02 in multivariable analysis) and OS (P = .007, log-rank test; P = .003 in multivariable analysis) compared with patients with a T/T genotype. These results suggest that patients with a predominant dependence on NKG2A–HLA-E interaction for NK cell regulation and education display lower relapse after HDC/IL-2 immunotherapy. We also investigated the potential benefit of the M/x genotype in a cohort of AML patients receiving fully matched allo-SCTs (ie, donor and recipient were matched with respect to their HLA-B −21 genotype). As shown in Figure 4C and D, there was no difference in outcome in terms of LFS (P = .49) or OS (P = .49) between the M/x and T/T genotypes in transplanted patients who had not received IL-2–based immunotherapy.
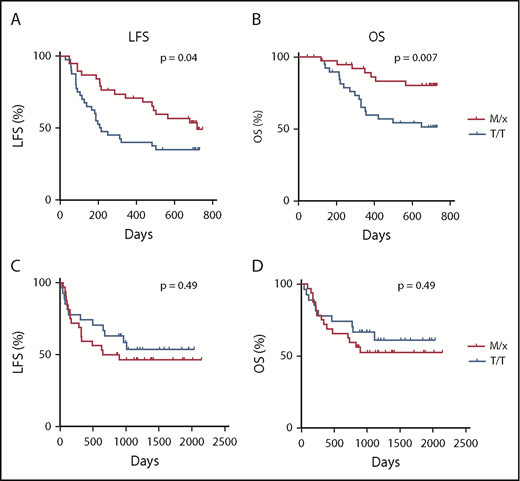
Impact of HLA-B −21 genotype on outcome of AML patients. LFS (A) and OS (B) for patients with an M/x genotype (n = 38) or T/T genotype (n = 42) receiving HDC/IL-2 treatment. Five patients (6%) had a M/M genotype, 33 patients (41%) had an M/T genotype, and 42 patients (53%) had a T/T genotype. LFS (C) and OS (D) for AML patients undergoing transplantation with an M/x (n = 32) or T/T (n = 27) genotype.
Impact of HLA-B −21 genotype on outcome of AML patients. LFS (A) and OS (B) for patients with an M/x genotype (n = 38) or T/T genotype (n = 42) receiving HDC/IL-2 treatment. Five patients (6%) had a M/M genotype, 33 patients (41%) had an M/T genotype, and 42 patients (53%) had a T/T genotype. LFS (C) and OS (D) for AML patients undergoing transplantation with an M/x (n = 32) or T/T (n = 27) genotype.
Discussion
The recently described dichotomy in NK cell regulation that depends on the HLA-B −21 genotype has been suggested to be of relevance for HIV control,20,22 but no study has addressed its potential role in the clinical outcome in malignancy. In this study, we found that AML patients with HLA-B −21M/x displayed superior LFS and OS compared with individuals with the HLA-B −21T/T genotype during IL-2–based immunotherapy. The superior outcome for individuals with NKG2A-regulated NK cells is intriguing, because we observed that NK cells recovered from AML patients already displayed a high proportion of NKG2A+ cells at the onset of therapy, which suggests that these cells could be inhibited by HLA-E. The high expression of NKG2A was not restricted to immature KIR− or single-KIR+ subsets, because NK cells with 2 or 3 of the major inhibitory KIRs were also found to coexpress NKG2A. The fraction of NKG2A-expressing cells increased further in all NK cell subsets in response to IL-2, regardless of whether they expressed 1 or 2 KIRs.
The aberrant NKG2A expression during HDC/IL-2 immunotherapy indicated that NKG2A was a marker for activation. A transcriptome analysis of NK cells during IL-2 stimulation showed that NKG2A transcripts increased during the first day of IL-2 stimulation in vitro. Interestingly, the transcript levels of inhibitory receptors normally associated with T cells, also known as immune checkpoint, displayed a similar pattern in NK cells. By contrast, the 3 major KIRs displayed reduced transcript levels after IL-2 stimulation, but we did not observe any reduction in KIR expression at the protein level in patients receiving HDC/IL-2 immunotherapy. An explanation for the lack of impact of NKG2A expression on clinical outcome may be that activated NK cells are able to override the inhibitory signals generated by NKG2A–HLA-E interactions. Although transduced 221-AEH cells expressing supraphysiological levels of HLA-E effectively inhibited NK cell degranulation, a T2 cell assay with adjustable HLA-E expression revealed that more physiological levels of HLA-E could inhibit degranulation by resting NK cells but not by IL-2–stimulated NK cells. In line with this finding, IL-2–activated NK cells overrode NKG2A signaling to a greater extent than KIR signaling in assays using KIR ligand–matched AML blasts.
The differential capacity of NKG2A and KIRs to withstand activated NK cells is interesting in view of the above-mentioned study by Horowitz et al,20 which postulated that human NK cells are predominantly educated, and regulated, by NKG2A or KIRs. Individuals with an HLA-B −21M/x genotype have slightly higher HLA-E expression; accordingly, NKG2A+ subsets from these donors were better educated and displayed stronger effector functions against HLA-deficient K562 cells than did cells from T/T donors. In agreement with the view that NKG2A-regulated NK cells are more prone to override the inhibitory signals controlling them, IL-2–stimulated NK cells from M/x donors displayed superior degranulation responses to KIR ligand–matched AML blasts. Notably, only the NKG2A+ NK cell subsets accounted for the increased degranulation response by M/x donors.
In contrast to the observed benefit of an M/x genotype in AML patients receiving HDC/IL-2 immunotherapy, no improved survival was observed in a cohort of AML patients receiving a fully matched allo-SCT. In the context of HDC/IL-2 immunotherapy, the therapy-induced upregulation of activating receptors may enable NK cells to override NKG2A signaling but not the stronger inhibitory KIR-mediated signals present in T/T patients. Along with this reasoning, the benefit of low expression of HLA-ABC in the myeloid compartment27 was predominantly observed in M/x patients; only in those patients may the reduction in inhibitory ligands be sufficient to overcome the inhibitory signals. Multiple studies have shown that NKG2A inhibition can be overridden by NK cells, either by signaling via KIR2DS1–HLA-C235 or by signaling engaging NKG2D and MICA/B,36 or in NK cells transduced with a CAR construct.18 The ability to override NKG2A, but not KIR, may be related to the intracellular organization of the inhibitory signaling complex. Although NKG2A is a type II membrane protein, with the C-terminal exposed to the exterior and the N-terminal located at the cytoplasmic side, inhibitory KIRs (iKIRs) are organized in the opposite manner.37 Accordingly, the 2 ITIMs in the intracellular portion of the receptors are oriented in opposite directions. For iKIRs, the more important N-terminal ITIM is located close to the membrane, whereas it is located more distally for NKG2A.38,39 Thus, it is possible that the opposite orientation of the ITIMs results in different capacities of NKG2A and iKIRs to signal via the N terminal ITIM or to engage both SH2 domains of SHP-1/2, which may potentially lead to a different capacity to transduce an inhibitory signal for the 2 receptors. An alternative explanation is given by the relative expression levels of different inhibitory ligands. HLA-A and HLA-B display ∼5 times higher expression levels than HLA-C, which, in turn, is expressed 25 times more than HLA-E.34 Thus, the ligand for NKG2A is relatively scarce and, therefore, may only generate limited inhibition to NKG2A+ cells.
The clinical consequences of the dimorphism in HLA-B −21 are largely unknown. Merino et al have shown that M/M partners of HIV-infected individuals will acquire the infection more rapidly23 and that the enhanced NKG2A-mediated inhibition affects the killing capacity of HIV-infected target cells.24 Furthermore, Ramsuran et al reported high HLA-A expression to be associated with high viral load in HIV-infected subjects and that the impact of HLA-A expression was preferentially observed in patients with HLA-B −21M alleles.22 The investigators proposed a chain of events by which a high supply of HLA-A leader peptides enhances expression of HLA-E to limit NK cell–dependent control of infected cells. At first glance, these results are contradictory to our observations; however, there are important differences between the disease states that need to be considered. During HIV infection, the cell surface expression of HLA molecules is altered in 2 ways: HLA expression is induced by the antiviral IFN response, and the HIV-encoded protein, Nef, downregulates HLA-A and HLA-B by inducing internalization of HLA, whereas other HLA class I molecules are spared.40 Thus, the translation and intracellular transport of membrane vesicles containing HLA through the endoplasmic reticulum and Golgi are unaffected by Nef,40 whereas the supply of leader sequences for HLA-E presentation is increased, leading to elevated HLA-E expression.41,42 Furthermore, the downregulation of HLA-A and HLA-B may allow missing self-recognition by KIR3DL1+ NK cells in Bw4+ patients. Thus, in HIV, the balance between the 2 inhibitory systems is altered; M/M individuals receive enhanced inhibitory input, whereas T/T individuals receive reduced inhibitory input, through KIR3DL1. Another factor to consider is that high-expressing HLA-C alleles are protective in HIV, because they allow development of efficient HLA-C–restricted T-cell responses.43,44 Transcripts of low-expressed HLA-C alleles are targeted by miR-148a, and the single-nucleotide polymorphism determining microRNA susceptibility is 1 of the major genetic determinants for outcome after HIV infection.43,45 Notably, the low-expressed HLA-C alleles are in strong linkage disequilibrium with HLA-B −21M20 ; thus, the observed disadvantage for M/M individuals with regard to HIV infection may be related, at least in part, to low expression of HLA-C.
AML is a highly heterogenous disease, and AML blasts may display differential expression of NK cell receptor ligands and susceptibility to NK cell cytotoxicity.46,47 In this study, we show that HLA-B −21M/x individuals, whose NK cells are primarily controlled by NKG2A–HLA-E interactions, had a more favorable outcome following HDC/IL-2 immunotherapy for AML than did T/T individuals. Our data suggest that the relatively low expression of HLA-E on AML blasts enables cytokine-stimulated NK cells to override NKG2A-dependent signaling to a larger extent than KIR-dependent signaling. However, it cannot be excluded that the impact of the HLA-B −21 dimorphism may be restricted to certain subtypes of AML (eg, low HLA-E expression and high susceptibility to NK cell cytotoxicity). Our results do not preclude a negative impact of NKG2A–HLA-E interactions on clinical outcome in certain subgroups of AML patients. A therapeutic antibody targeting NKG2A, monalizumab, is under investigation in various malignancies, including leukemia (NCT02921685). The upregulation of NKG2A during HDC/IL-2 immunotherapy suggests that there may be a rationale for combining HDC/IL-2 and NKG2A blockade to further improve the outcome for certain subgroups of AML. Nevertheless, it should be noted that HLA-B −21 M/x was an independent predictor of LFS and OS in a multivariable Cox regression analysis of the AML patient cohort receiving HDC/IL-2 immunotherapy.
In summary, our results demonstrate that a dimorphism in HLA-B impacts NK cell responses toward leukemic blasts, which translates into a more favorable clinical outcome for HLA-B −21M/x AML patients receiving IL-2–based immunotherapy.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by the Swedish Research Council, the Swedish Cancer Society, the Swedish state via the ALF agreement, the Clas Groschinskys Foundation, the Åke Wiberg Foundation, the Assar Gabrielsson Foundation, the Lion Cancer Foundation West, the Wilhelm and Martina Lundgren Research Foundation, BioCARE, and the Sahlgrenska Academy at the University of Gothenburg.
Authorship
Contribution: A.H., E.B., K.H., and F.B.T. designed the research; A.H., E.B., F.E.S., and B.A.H. performed research and analyzed data; M.B. contributed patient material; J.A., A.M., K.H., and F.B.T. supervised the study; and all authors wrote and reviewed the manuscript.
Conflict-of-interest disclosure: K.H., J.A., A.M., F.E.S., and F.B.T. are authors of issued or pending patents protecting the use of HDC in cancer immunotherapy. The remaining authors declare no competing financial interests.
Correspondence: Fredrik B. Thorén, University of Gothenburg, Box 425, 40530 Gothenburg, Sweden; e-mail: fredrik.thoren@gu.se.

![Figure 1. NKG2A and granzyme B distribution and responses of NK cells in vivo and in vitro. (A) Distribution of NK cell subsets in AML patients before treatment start (n = 64) and in healthy donors (n = 24). Frequency of NKG2A+ NK cells in subsets expressing 0, 1, 2, or 3 KIRs in AML patients (n = 54) before or after cycle 1 (B) and in unstimulated and IL-2–stimulated (500 U/ml) healthy donors (n = 6) (C). (D) Fold change, compared with start, in messenger RNA expression of NKG2A after IL-2 stimulation. (E) Granzyme B expression (median fluorescence intensity [MFI]) in unstimulated and IL-2–stimulated NK cell subsets (n = 24). (F) Percentage increase in granzyme B response between unstimulated and IL-2–stimulated NK cells (n = 24). (G) LFS for patients dichotomized by median NKG2A expression at treatment start (low, n = 29 or high, n = 30). (H) LFS for patients dichotomized by median frequency of NKG2A+ NK cells (low, n = 29 or high, n = 30). *P < .05, **P < .005, ***P < .0005.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/13/10.1182_blood-2018-09-874990/3/m_blood874990f1.png?Expires=1768877998&Signature=ZuY-rdxbXcw1CsGxFBmohVE6IgvldUW6ed3m9ve4vT2~aoORyeLd0AfMvdKOljfxCCeqiv-6RklFyPjNN01fYWCJVhtQ75yFZiw~o0QZTJjv8RNF-NplykJc0everGx023xYjo8LZoBu3z~UEjGKn6PEt-ojAyTpr1BJbcVVyFGYtR4v-OFsenqgPwMzpIZ2ExmoTla7RItesl-t89tL0mhtRMZHJanpIOX8KodCxIINNk49y6N-hwFlHwWUMes-hhuoHouzffZgizCvWo4nRowxPOPAqR6UxQbqgW5360saDaZot7SckmQoEg83~hADJMW~z1ynGybMSWktZgDaZw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)