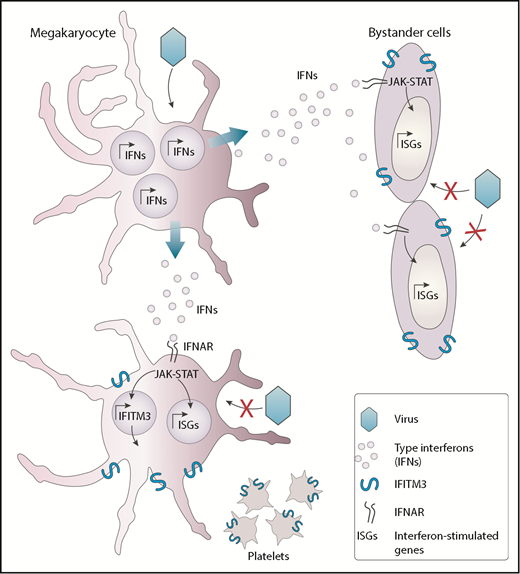
Megakaryocytes (MKs) are mostly recognized for their role in the generation of platelets. In this issue of Blood, demonstrate that MKs are also capable of preventing the spread of viral infection, suggesting that they too participate in immunity.1
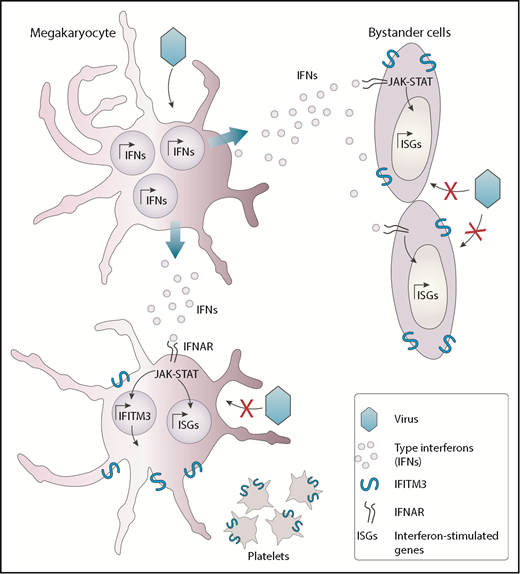
MKs possess intrinsic antiviral immunity through regulated induction of IFITM3. Viruses such as dengue virus that can infect MKs trigger the production of interferon molecules. The released interferons (IFNs) interact with its receptor (IFNAR) and alarm neighboring cells, such as other MKs and bystander cells from other lineages, thereby inducing a series of interferon-stimulated genes (ISGs), including IFITM3. This response participates in the establishment of a potent antiviral state that protects cells from incoming viral infections.
MKs possess intrinsic antiviral immunity through regulated induction of IFITM3. Viruses such as dengue virus that can infect MKs trigger the production of interferon molecules. The released interferons (IFNs) interact with its receptor (IFNAR) and alarm neighboring cells, such as other MKs and bystander cells from other lineages, thereby inducing a series of interferon-stimulated genes (ISGs), including IFITM3. This response participates in the establishment of a potent antiviral state that protects cells from incoming viral infections.
Type I interferons, which include α and β interferons, are among the most effective soluble antiviral mediators secreted in response to an infecting virus. Although interferon-α is mainly secreted by leukocytes, with plasmacytoid dendritic cells being the penultimate producing cell type, interferon-β can be secreted by cells of many origins. Once secreted, interferons act to alert nearby cells that an intruder is present. On type I interferon receptor engagement and kinase cascade signaling, several dozen genes are rapidly turned on, leading to an antiviral state that will interfere with virus replication and restrict infection by incoming viruses.
Whether MKs could respond to virus infection and alert other cells was, however, unknown until now. The authors have identified that in response to systemic and pulmonary viral infections mediated by dengue and influenza, platelets overexpress the interferon-induced transmembrane 3 (IFITM3) restriction factor. Considering that the platelet content is derived from MKs, it is presumed that type I interferons secreted in response to these viruses first activate IFITM3 in MKs, which is then passed on to platelets. Using a variety of compelling experimental models, including human subjects infected or vaccinated with dengue or influenza viruses, the authors provide convincing evidence that MKs respond to natural viral infections by secreting interferons and upregulating IFITM3. Secreted interferons can act in a paracrine manner to induce an antiviral state in neighboring cells, thereby controlling the spread of the infection (see figure). Such antiviral biological activities were never reported and add to recently identified immune functions of MKs such as interleukin-1 production in arthritis and major histocompatibility complex class I antigen presentation.2,3
Although recent studies have questioned the appropriateness of certain mouse models to examine the contribution of platelets in immunity resulting from pf4-Cre transgene expression in certain leukocyte populations,4 a major strength of the present study is the use of clinically relevant human biospecimens. The antiviral gene identified by the authors, IFITM3, was consistently overexpressed in the platelets of human subjects naturally infected with dengue or influenza virus. IFITM3 is a 133-amino acid protein that disrupts cholesterol homeostasis, thus inhibiting the entry of several viruses (including influenza and dengue viruses) to the host cell cytoplasm by preventing fusion of the viral envelope with the endosome.5 Of interest, compared with severely affected patients, individuals with milder symptoms of dengue infection were the ones with the greatest plasmatic levels of interferon-α and with the more robust platelet IFITM3 expression. Although dengue viral loads were not reported in the study, one would expect an inverse correlation between interferon-α levels and viral loads. Similarly, when analyzing patients infected with H1N1 influenza infection, survivors were those who demonstrated a gradual increase in platelet IFITM3 expression over time. These results clearly show that platelets have increased IFITM3 protein expression during acute viral infections. To demonstrate that MKs were the ones responding to viral infections, the authors derived human MKs from CD34+ cells and infected them in vitro with dengue virus. In response to infection, MKs secrete interferons that turned on several genes, including IFITM3. The importance of IFITM3 as a dengue restriction factor was further supported by a populational study, indicating that human subjects carrying a loss of function in IFITM3 resulting from a homozygous mutation (rs12252-C) are significantly more susceptible to dengue infection. The authors were also able to demonstrate that MKs carrying the rs12252-C mutation showed increased permissiveness to dengue virus replication, highlighting the importance of IFITM3 in MK resistance to viral infection.
Using a live-attenuated dengue vaccine, the authors were able to demonstrate that 14 days postvaccination, levels of platelet IFITM3 were increased, suggesting that replication of the attenuated virus was sufficient to trigger the release of type I interferons leading to IFITM3 gene activation. In contrast, no increase in platelet IFITM3 levels were observed 14 days after vaccination with inactivated trivalent influenza vaccine, arguing that viral replication might be needed to induce sufficient levels of interferons to achieve a systemic effect. It should be kept in mind, however, that inactivated trivalent influenza vaccines do induce an interferon response, with maximal effects observed within 3 days postvaccination and return to baseline by 7 days.6
A commonly observed consequence of dengue virus infection is thrombocytopenia. The underlying mechanism is currently unknown, but the authors have provided convincing evidence that dengue virus can directly infect MKs, likely affecting platelet production and possibly contributing to thrombocytopenia. Furthermore, although platelets are anucleated and not typically considered prime target cells of viral infection, Simon et al reported that platelets do get infected and are capable of replicating all 4 dengue serotypes.7 Knowing that MKs and platelets both express IFITM3 in response to interferon stimulation, it is thus likely that the mechanism revealed in the present study is relevant to antidengue defense. Whether platelets overexpressing IFITM3 are resistant to dengue or other virus infection/replication would provide additional biological significance to the current work. Such findings would be especially relevant for pulmonary viral infections, considering that lungs are an important site of terminal platelet production.8
Conflict-of-interest disclosure: The authors declare no competing financial interests.