

In this study, 1 (in the research group of Andre Larochelle) shed light on the mechanism of pancytopenia in inflammation, a long-standing clinical observation lacking an explanation. Interferon-γ (IFN-γ), a mediator of the inflammatory response, decreases hematopoietic stem and progenitor cell (HSPC) function by binding to thrombopoietin (TPO), thereby preventing it from signaling via its own receptor, c-MPL. This same understanding provides insight into how the TPO mimetic eltrombopag (ELT) can overcome IFN-γ–mediated inhibition of hematopoiesis, because it is unaffected by IFN-γ binding and, thus, can engage c-MPL and induce downstream signaling.
Prior to this study, it was well established that chronic inflammation can cause pancytopenia. Elevated levels of IFN-γ associated with multiple types of inflammation can inhibit HSPC function, leading to neutropenia and thrombocytopenia. IFN-γ plays a role in the induction of acquired aplastic anemia and anemia of inflammation.2 TPO mimetics, such as ELT and romiplostim, are being used successfully in the treatment of patients with bone marrow failure and, in particular, aplastic anemia, despite elevated levels of endogenous TPO.3-5 Megakaryocytes, as well as HSPCs, express c-MPL, and TPO is 1 of the major cytokines maintaining hematopoiesis. Lack of functional c-MPL or TPO (eg, due to mutations in c-MPL in patients with congenital amegakaryocytic thrombocytopenia) eventually results in pancytopenia with transfusion dependence and risk of infections.
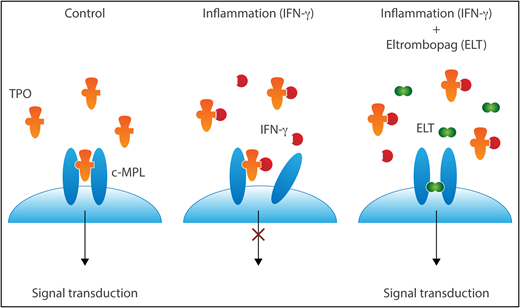
TPO signals by binding to and changing the conformation of c-MPL dimers. In the presence of IFN-γ, TPO cannot dimerize c-MPL. In the face of IFN-γ–mediated inhibition of c-MPL binding, ELT can still promote signaling via c-MPL dimers. Professional illustration by Somersault18:24.
TPO signals by binding to and changing the conformation of c-MPL dimers. In the presence of IFN-γ, TPO cannot dimerize c-MPL. In the face of IFN-γ–mediated inhibition of c-MPL binding, ELT can still promote signaling via c-MPL dimers. Professional illustration by Somersault18:24.
The elegant study by Alvarado et al provides a clear and intriguing explanation for the clinical scenario and its successful treatment with TPO mimetics, as represented in the simple model (see figure).
Knowing that ELT can overcome the inhibitory effects of IFN-γ on HSPCs, the investigators use in vitro and in vivo models to explore the mechanisms by which ELT rescues HSPC function. They set the stage for their studies by demonstrating that IFN-γ inhibits CD34 cell growth in vitro and provide phenotypic and functional data definitively proving that ELT can overcome this inhibition. They determine that IFN-γ abrogates TPO-induced signal transduction but not ELT-induced signal transduction. This suggests that, in the presence of IFN-γ, ELT can activate c-MPL, whereas TPO cannot, thereby addressing the previous conundrum as to why ELT is beneficial in patients who already have elevated levels of endogenous TPO.
Because TPO and ELT are known to bind to distinct sites on c-MPL, the investigators hypothesized that IFN-γ must prevent binding of TPO, but not ELT, to c-MPL. Interestingly, they found exactly this but with a twist. It was already known that TPO has 2 distinct cMPL interaction sites: 1 high affinity and 1 low affinity. TPO binding and activation of cMPL represent a 2-step process: TPO’s high-affinity site interacts with a first c-MPL subunit, followed by engagement of a second c-MPL subunit with TPO’s low-affinity site, thereby bringing c-MPL homodimers into close apposition and inducing conformational changes required for signal transduction. Using protein: protein association studies, they show that IFN-γ directly binds to TPO and specifically prevents the second stage of TPO binding to c-MPL. TPO–IFN-γ heterodimers can still engage with cells because the IFN-γ binding to TPO does not affect the higher-affinity binding to c-MPL. The TPO–IFN-γ complexes only block the weaker receptor-binding site of TPO, preventing homodimerization of c-MPL and the receptor conformational changes required to transduce intracellular signaling. How then does ELT overcome this inhibition? ELT has no structural similarities to TPO and is a unique nonpeptide TPO “mimetic” that activates c-MPL signaling via binding within the transmembrane domain. ELT is not recognized by IFN-γ; thus, it can induce c-MPL signaling, despite the presence of IFN-γ. It is also unaffected by high levels of TPO–IFN-γ complexes, because it binds a distinct region within c-MPL.
Why then does ELT not fully overcome IFN-γ effects, and why is it not effective in every patient? IFN-γ also signals HSPCs directly via IFN receptors to promote expression of SOCS proteins, which can inhibit TPO/c-MPL signaling downstream of receptor activation.6 Indeed, as expected, ELT cannot overcome IFN-γ–mediated upregulation of SOCS proteins, limiting its effect to direct activation of c-MPL.
This study leads to many important questions that will surely be pursued in the future. Can one detect TPO–IFN-γ heterodimers in patients with inflammation-associated pancytopenia and elevated TPO levels? What is the structural basis for TPO–IFN-γ binding? Can TPO–IFN-γ binding be targeted and exploited therapeutically? Are other molecules bound by TPO and IFN-γ that may modulate this interaction/effect?
In summary, this study provides the mechanistic basis for the well-known finding that inflammation can result in pancytopenia and the previously discovered clinically beneficial effects of ELT in patients with immune-mediated aplastic anemia, despite high endogenous levels of TPO. The data suggest that ELT will be beneficial in other cases of bone marrow failure associated with inflammation and elevated levels of IFN-γ, such as in chronic infection, graft-versus-host disease, and autoimmunity.
Conflict-of-interest disclosure: D.S.K. declares no competing financial interests.

