

Key Points
Excess IFN-γ perturbs TPO-induced signaling pathways in human HSPCs; eltrombopag enhances HSPC function by overcoming this inhibition.
Steric occlusion of the low-affinity binding site of TPO to c-MPL by TPO:IFN-γ heteromers contributes to perturbation of TPO signaling.

Abstract
The proinflammatory cytokine interferon-γ (IFN-γ) has been implicated in human hematopoietic stem and progenitor cell (HSPC) depletion in immune-mediated bone marrow failure syndromes. We show that IFN-γ specifically prevents full engagement of thrombopoietin (TPO), a primary positive regulator of HSPC survival, to its receptor (c-MPL) via steric occlusion of the low-affinity binding site, contributing to perturbation of TPO-induced signaling pathways and decreased survival of human HSPCs. Eltrombopag, a synthetic small molecule mimetic of TPO that interacts with c-MPL at a position distinct from the extracellular binding site of TPO, bypasses this inhibition, providing an explanation for its clinical activity in bone marrow failure, despite already elevated endogenous TPO levels. Thus, IFN-γ–mediated perturbation of TPO:c-MPL complex formation and the resulting inhibition of a critical pathway of growth factor cell signaling may represent a general mechanism by which IFN-γ impairs the function of human HSPCs. This understanding could have broad therapeutic implications for various disorders of chronic inflammation.
Introduction
Chronic exposure to inflammatory cytokines is associated with numerous diseases in humans. Interferon-γ (IFN-γ) is a pleiotropic mediator of inflammation and immunity. Inhibition of hematopoiesis by IFN-γ has been reported in assays of human progenitor cells in vitro,1-6 in in vivo murine studies,7-10 and by inference from clinical observations.1,11,12 Aplastic anemia (AA), the archetypical human bone marrow (BM) failure syndrome, is a T-cell–mediated autoimmune disorder resulting, at least in part, from the suppressive effects of Th1 cytokines, primarily IFN-γ.13-15 Hematopoietic stem and progenitor cells (HSPCs) are greatly reduced in patients with severe AA. If left untreated, the associated pancytopenia causes life-threatening infections, bleeding, and anemia, and most patients die within 1 year after diagnosis. Approximately two thirds of subjects respond to a single course of immunosuppressive therapy (IST) with anti-thymocyte globulin and cyclosporine.16-18 However, attempts at modulating the immune response further have failed to increase the rate of response,19-21 likely due to the stem cell deficit left untreated by IST alone in the most affected patients.
In a recent study, the negative impact of IFN-γ on HSPC proliferation was attributed to IFN-γ–mediated perturbation of a critical pathway of cell signaling activated by the hematopoietic cytokine thrombopoietin (TPO).9 Together with its receptor, c-MPL, TPO acts as the primary regulator of HSPC survival.22 The importance of the TPO:c-MPL axis in early hematopoiesis was initially elucidated in murine HSPCs.23-28 The c-MPL receptor was shown to be expressed on these cells, and significant proliferation was observed ex vivo in the presence of TPO and other cytokines. Transgenic mice lacking c-MPL or TPO,23,24,27 and children born with loss of functional mutations in c-MPL or TPO,29-33 develop progressive pancytopenia due to a reduction in HSPC numbers over time.
Analysis of TPO’s crystal structure demonstrated that it interacts with the extracellular region of c-MPL in a 1:2 stoichiometry, with 1 high-affinity and 1 low-affinity binding site.34 Upon binding of TPO, receptor homodimerization and/or stimulation of preformed inactive dimers is promoted, leading to global receptor conformational changes that are translated through the transmembrane (TM) and cytosolic domains. Residues of the cytosolic domain are phosphorylated in trans by JAK2 kinases, triggering signal transduction cascades, primarily the JAK-STAT3/5, PI3K/AKT, and ERK/MAPK pathways.22 TPO’s functional effects are regulated by internalization and degradation of the activated c-MPL receptor,35 as well as negative regulatory signals largely induced by the suppressor of cytokine signaling (SOCS) family of proteins.36,37 Upregulation of SOCS proteins by IFN-γ has been proposed as a possible mechanism for IFN-γ interference with TPO signaling in HSPCs.9
Eltrombopag is a synthetic orally bioavailable nonpeptidyl small molecule mimetic of TPO that binds to the TM domain of c-MPL. In recent clinical trials, eltrombopag was found to improve trilineage hematopoiesis in patients with AA refractory to IST38,39 and, when combined with IST in previously untreated patients, eltrombopag was associated with higher rates of hematologic response than observed historically.40 Improved BM indices and trilineage hematologic responses provided evidence that eltrombopag likely acts by stimulating the small number of residual HSPCs in the BM.
Paradoxically, endogenous TPO levels are already markedly elevated in patients with severe AA compared with healthy individuals or subjects with immune thrombocytopenic purpura.41,42 Other hematopoietic cytokines, such as granulocyte colony-stimulating factor (G-CSF) and erythropoietin (EPO), are also elevated in severe AA, but adjunct therapy with these growth factors is not efficacious. Therefore, the mechanism by which eltrombopag could promote hematopoiesis and improve the stem cell deficit in the setting of elevated TPO levels was a conundrum. We hypothesized that eltrombopag might support HSPC survival by activating signaling pathways in these cells distinctly from endogenous TPO under inflammatory conditions. Here, we demonstrate IFN-γ–mediated disruption of TPO:c-MPL complexes as a molecular mechanism to explain the suppressive effects of IFN-γ in human HSPCs, and we show that eltrombopag bypasses this inhibition in BM failure syndromes.
Methods
See supplemental Methods (available on the Blood Web site) for a full description of methods.
Human CD34+ HSPCs
Human CD34+ HSPCs were obtained from healthy male and female volunteers (aged 18-56 years) after informed consent, in accordance with the Declaration of Helsinki, under an Institutional Review Board–approved clinical protocol (NCT00001529). Patients underwent mobilization with a daily subcutaneous injection of 10 μg/kg G-CSF (filgrastim; Amgen) for 5 days, followed by leukapheresis. CD34+ HSPCs were enriched using a CliniMACS Plus Instrument (Miltenyi Biotec) and cryopreserved prior to culture.
Cell culture and cytokines
Human CD34+ HSPCs were thawed and resuspended in StemSpan Serum-Free Medium II (STEMCELL Technologies) supplemented with 5 ng/mL human stem cell factor (SCF) and FMS-like tyrosine kinase 3 ligand (Flt3L) and 3 to 5 ng/mL recombinant human TPO (TPO), 3 to 5 ng/mL TPO + 100 ng/mL IFN-γ, 3 µg/mL eltrombopag, 3 µg/mL eltrombopag + 100 ng/mL IFN-γ, or 100 ng/mL IFN-γ. All growth factors were purchased from PeproTech. Cells were cultured at 37°C under standard conditions for 7 to 14 days.
Signaling–phosphoflow experiments
Human CD34+ HSPCs were resuspended in stimulation medium that consisted of StemSpan medium supplemented with the previously mentioned cytokines. A culture medium control was used at each time point for each donor to account for the variable background fluorescence observed among experiments, donors, and batches of medium. Cells from all conditions were incubated at 37°C for a predetermined period of time ranging from 5 minutes to 4 hours. Next, cells were fixed, permeabilized, and stained with antibodies against phosphorylated (p)STAT5, pSTAT3, pAKT, or pERK1/2 (BD Biosciences) for 1 hour at room temperature. All stained cells were analyzed using an LSR II Fortessa flow cytometer (BD Biosciences), and mean fluorescence intensity (MFI) was calculated as the MFI of each condition minus the MFI of StemSpan alone for that same time point. To underscore the impact of IFN-γ and the observed differences in the magnitude of stimulation between TPO and eltrombopag within each pathway, MFI percentages were calculated by dividing the MFI stimulated by TPO or eltrombopag, alone or in combination with IFN-γ, at each time point within a specific signaling pathway, by the maximum MFI measured within that pathway. Signaling pathways were considered activated by a specific cytokine when MFI percentages were >10%.
Colony-forming unit assay
CD34+ HSPCs were expanded for 7 days under the cell culture conditions described above. On the final day of culture, cells were collected and cultured in duplicate for 11 days in methylcellulose-based medium at a concentration of 300 cells per milliliter (MethoCult H4435 Enriched; STEMCELL Technologies). On day 11, total colony-forming units (CFU) were quantified using an EVOS XL Core transmitted-light microscope (Thermo Fisher Scientific) and normalized to the total number of expanded cells by donor and condition.
NOD-Scid-IL2rγnull mouse repopulation assay
Sublethally irradiated (270 cGy) 6- to 12-week-old female NOD-Scid-IL2rγnull (NSG) mice were transplanted by tail vein injection with the expanded progeny of human CD34+ cells that were cultured for 7 days as described above. BM cells were collected 12 weeks posttransplantation and stained with anti–human CD45 conjugated to FITC, anti-human CD20-phycoerythrin (PE), and anti-human CD13-PE. Animals were housed and handled in accordance with the guidelines set by the Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (DHHS publication NIH 85-23), and the protocol was approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute (NHLBI).
Microscale thermophoresis
Binding between TPO and c-MPL and binding between TPO and IFN-γ, SCF, or Flt3L were characterized using microscale thermophoresis (MST). Briefly, all reactions were incubated at room temperature for ≥1 hour prior to performing MST measurements. The reaction buffer for all experiments was 50 mM Na3PO4 pH 6.0, 100 mM NaCl, and 0.05% Tween-20. Measurements were performed in premium-treated capillaries on a Monolith NT.115 system (both from NanoTemper Technologies) using 50% infrared laser power and 99%, 95%, or 75% LED intensity with 10, 20, or 50 nM labeled protein, respectively. Laser on and off times were set at 30 seconds and 5 seconds, respectively. Data were acquired and analyzed using MO.Control and MO. Analysis softwares (NanoTemper Technologies), respectively.
Multispectral imaging flow cytometry–ImageStream System
Human CD34+ cells were stained with mouse anti-human CD34-PE (BD Pharmingen) and CD110–Brilliant Violet 421 (BD Horizon), per the manufacturer’s recommendations, prior to the exposure to cytokines. Cell samples were fixed with 2% paraformaldehyde at 5, 15, 30, 45, and 60 minutes after stimulation. Using Inspire data acquisition software (Amnis), images of 10 000 CD34+ cells were captured on channel 1 for brightfield; channel 3 for PE, representing CD34 staining as an internalization-incapable control protein; and channel 7 for Brilliant Violet 421, representing c-MPL staining. Excitation of the samples was performed with 488-nm and 561-nm lasers at power settings of 120 mW and 200 mW, respectively. Data analysis was performed using IDEAS software (Amnis).
Fluorescence spectroscopy
The fluorescence spectrum of eltrombopag, an intrinsically fluorescent molecule, was measured using Qubit 3.0 (Thermo Fisher Scientific). The “blue (470 nm)” fluorescent mode was selected, which excites the solution at 470 nm and captures the emission spectrum between 510 and 580 nm and between 665 and 720 nm. We report the maximum fluorescence intensity in the latter window. Eltrombopag was held at a constant concentration of 100 µM and titrated with 0.1, 0.5, and 1.0 µM IFN-γ. All reactions were incubated for ≥1 hour at room temperature prior to performing fluorescence measurements. The reaction buffer for all experiments was 50 mM Na3PO4 pH 6.0, 100 mM NaCl, and 0.05% Tween-20. The intrinsic fluorescence of the reaction buffer was also measured and reported.
Results
Eltrombopag maintains human HSPCs in the presence of IFN-γ ex vivo
To address why eltrombopag is efficacious, despite high endogenous TPO levels in severe AA, we first cultured human CD34+ HSPCs from 11 healthy donors with and without IFN-γ and then compared the relative effect of TPO and eltrombopag on cell survival and function (Figure 1A). Ex vivo culture of normal CD34+ cells with IFN-γ has previously been shown to dependably model the impact of T-cell attack on marrow observed in AA patients.43 When CD34+ cells were cultured for 7 days in TPO-containing medium, addition of IFN-γ caused a significant reduction (2.8-fold) in the percentages and numbers of CD34+ cells relative to cultures without IFN-γ. In contrast, in cultures supplemented with an equivalent concentration of eltrombopag as a substitute for TPO (supplemental Figure 1), IFN-γ produced a much lower (1.4-fold) decrease in CD34+ cell percentages and absolute counts measured at 7 days compared with cultures with no IFN-γ (Figure 1B-C). The improvement in numbers of CD34+ cells with eltrombopag relative to TPO in the presence of IFN-γ was due to reduced cell death in the presence of eltrombopag; there were no significant differences in lineage differentiation or cell proliferation (supplemental Figure 2).
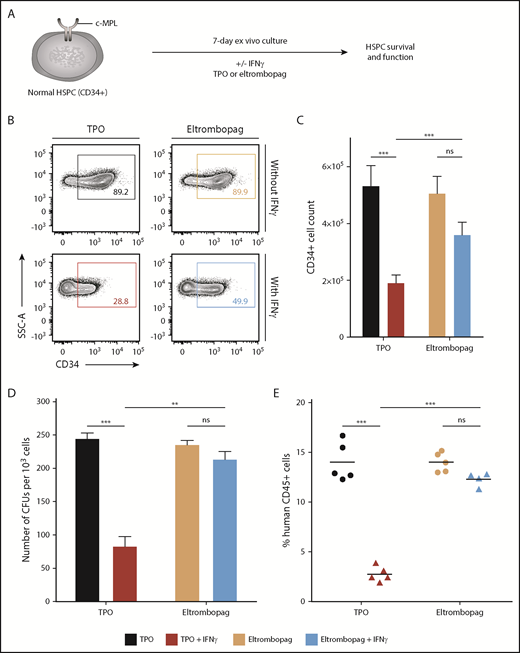
Eltrombopag maintains human HSPCs in the presence of IFN-γ in vitro. (A) Schematic of the experimental design. (B) Representative flow cytometry plots of CD34 expression obtained after 7 days of culture. (C) Absolute CD34+ cell counts after 7 days of culture (n = 13, from 11 independent donors). (D) Normalized numbers of CFU derived from the 7-day cultures (n = 3 independent donors). (E) Human cell engraftment, as measured by human CD45-expressing cells in the BM of recipient NSG mice 12 weeks after transplantation of human CD34+ cells cultured for 7 days (n = 4-5 mice per group). Human lymphoid (CD20+) and myeloid (CD13+) cells were detected in all animals. (C-E) Data are mean ± standard error of the mean (SEM). **P < .01, ***P < .001, multiple 2-tailed unpaired Student t tests. ns, nonsignificant.
Eltrombopag maintains human HSPCs in the presence of IFN-γ in vitro. (A) Schematic of the experimental design. (B) Representative flow cytometry plots of CD34 expression obtained after 7 days of culture. (C) Absolute CD34+ cell counts after 7 days of culture (n = 13, from 11 independent donors). (D) Normalized numbers of CFU derived from the 7-day cultures (n = 3 independent donors). (E) Human cell engraftment, as measured by human CD45-expressing cells in the BM of recipient NSG mice 12 weeks after transplantation of human CD34+ cells cultured for 7 days (n = 4-5 mice per group). Human lymphoid (CD20+) and myeloid (CD13+) cells were detected in all animals. (C-E) Data are mean ± standard error of the mean (SEM). **P < .01, ***P < .001, multiple 2-tailed unpaired Student t tests. ns, nonsignificant.
In human progenitor assays, normalized numbers of CFU derived from the 7-day cultures containing IFN-γ decreased threefold in TPO-supplemented medium but remained mostly unchanged in the presence of eltrombopag, relative to cultures without IFN-γ (Figure 1D). To compare the impact of IFN-γ exposure on HSC function in cultures containing TPO or eltrombopag, we transplanted 7-day expanded progeny of an equal starting number of CD34+ cells for each culture condition into NSG mice. In TPO-containing cultures, CD34+ cells exposed to IFN-γ showed a marked engraftment deficit relative to cells cultured without IFN-γ. Notably, human cell engraftment was nearly identical across all mice transplanted with CD34+ cells cultured in eltrombopag-supplemented medium, irrespective of IFN-γ exposure (Figure 1E). Together, these data indicate that eltrombopag has a distinct ability to maintain human HSPCs under inflammatory conditions in an ex vivo model of BM failure.
Eltrombopag bypasses IFN-γ blockade of TPO intracellular signaling in human HSPCs
To understand the potential mechanism by which eltrombopag allowed maintenance of human HSPCs in the presence of IFN-γ, we examined the impact of IFN-γ on intracellular signaling pathways induced upon binding of TPO and eltrombopag to c-MPL in CD34+ cells. Because little is known about signaling pathways evoked by TPO and eltrombopag in human CD34+ cells, we systematically measured induction of phosphorylation of STAT5, STAT3, AKT, and ERK at various times after treatment of these cells with either compound. TPO and eltrombopag activated the JAK-STAT5 pathway, but with different kinetics. TPO induced a rapid and high-potency increase in STAT5 phosphorylation, followed by a rapid decay in signal, whereas eltrombopag caused a slow lower-potency increase in STAT5 phosphorylation, and the signal persisted longer. We also observed that STAT3 was only minimally phosphorylated in the presence of TPO, and the AKT signaling cascade was not stimulated by eltrombopag in these cells (supplemental Figure 3).
Next, we assessed the effect of IFN-γ exposure on the most activated signaling pathways for TPO and eltrombopag over a period of 15 minutes up to 4 hours. In CD34+ cells from 3 to 6 independent donors, IFN-γ markedly impaired TPO signaling, as measured by decreased phosphorylation of STAT5, AKT, and ERK (Figure 2A-C). Notably, eltrombopag-induced STAT5, STAT3, and ERK phosphorylation was completely preserved or even slightly increased in the presence of IFN-γ (Figure 2D-F). Hence, eltrombopag’s ability to bypass IFN-γ blockade of a critical pathway of growth factor cell signaling might explain its efficacy in maintaining HSPCs under inflammatory conditions in ex vivo cultures and in patients with immune-mediated marrow failure.
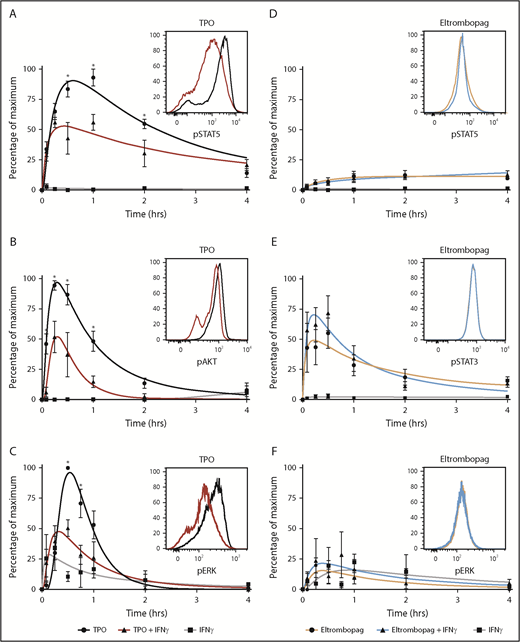
Eltrombopag bypasses IFN-γ blockade of TPO intracellular signaling in human HSPCs. The phosphorylation state of STAT5, STAT3, AKT, and ERK was measured using phosphoflow at various times after treatment of human CD34+ cells with TPO (A-C) or eltrombopag (D-F) in the presence or absence of IFN-γ. (A) JAK-STAT5 pathway for TPO (n = 5). (B) AKT pathway for TPO (n = 5). (C) ERK pathway for TPO (n = 3). (D) JAK-STAT5 pathway for eltrombopag (n = 5). (E) JAK-STAT3 pathway for eltrombopag (n = 6). (F) ERK pathway for eltrombopag (n = 3). Insets are representative flow cytometry graphs from 1 donor for each signaling pathway. Results at each time point are displayed as percentages of maximum ± SEM in individual pathways. To underscore the impact of IFN-γ and the observed differences in magnitude of stimulation within each pathway between TPO and eltrombopag, percentages of maximum were calculated by dividing the MFI stimulated by TPO or eltrombopag, alone or in combination with IFN-γ, at each time point within a specific signaling pathway, by the maximum MFI measured within that pathway. Curve fit by polynomial nonlinear regression. *P < .05, 2-way ANOVA.
Eltrombopag bypasses IFN-γ blockade of TPO intracellular signaling in human HSPCs. The phosphorylation state of STAT5, STAT3, AKT, and ERK was measured using phosphoflow at various times after treatment of human CD34+ cells with TPO (A-C) or eltrombopag (D-F) in the presence or absence of IFN-γ. (A) JAK-STAT5 pathway for TPO (n = 5). (B) AKT pathway for TPO (n = 5). (C) ERK pathway for TPO (n = 3). (D) JAK-STAT5 pathway for eltrombopag (n = 5). (E) JAK-STAT3 pathway for eltrombopag (n = 6). (F) ERK pathway for eltrombopag (n = 3). Insets are representative flow cytometry graphs from 1 donor for each signaling pathway. Results at each time point are displayed as percentages of maximum ± SEM in individual pathways. To underscore the impact of IFN-γ and the observed differences in magnitude of stimulation within each pathway between TPO and eltrombopag, percentages of maximum were calculated by dividing the MFI stimulated by TPO or eltrombopag, alone or in combination with IFN-γ, at each time point within a specific signaling pathway, by the maximum MFI measured within that pathway. Curve fit by polynomial nonlinear regression. *P < .05, 2-way ANOVA.
SOCS expression is equally upregulated by TPO and eltrombopag in the presence of IFN-γ
To elucidate the ability of eltrombopag to stimulate hematopoiesis in the setting of inflammation, we sought to characterize the mechanism by which the TPO mimetic overcame IFN-γ blockade of c-MPL signaling in human HSPCs. Because activation of c-MPL and IFN-γ receptors by their respective ligands is primarily regulated by negative-feedback mechanisms from the SOCS family of proteins,36,37 we queried whether TPO induced more SOCS-driven negative feedback than did eltrombopag in the presence of IFN-γ. Expression of the foremost members of the SOCS family (SOCS1, SOCS2, SOCS3, and CISH) was compared in CD34+ cells cultured with TPO or eltrombopag, in the presence or absence of IFN-γ. We found that expression of all tested SOCS proteins was equally upregulated by IFN-γ in combination with TPO or eltrombopag, as measured by quantitative reverse-transcription polymerase chain reaction and western blotting (supplemental Figure 4). Thus, although SOCS proteins likely contribute to the observed IFN-γ–induced perturbation of TPO signaling, we reasoned that there was an additional component to the mechanism, given eltrombopag’s ability to escape inhibition despite equivalent SOCS upregulation.
IFN-γ disrupts the low-affinity binding interaction between TPO and c-MPL
Eltrombopag binds c-MPL at a position distinct from the extracellular binding site of TPO, within the TM domain of the receptor44,45 ; thus, it cannot compete with TPO for binding. To clarify the mechanism of IFN-γ–mediated blockade of TPO signaling from c-MPL, we hypothesized that IFN-γ might impair receptor binding affinity of TPO but not eltrombopag. We used MST to assess the effect of IFN-γ on TPO binding to its receptor. MST is a technology used to determine binding affinities between macromolecules based on their directed movement along temperature gradients in solution (supplemental Figure 5A-C).46 Other techniques that are often used to study molecular interactions, such as surface plasmon resonance and isothermal titration calorimetry, were impractical because of the elevated nonspecific surface binding of TPO, a highly hydrophobic protein. MST utilizes hydrophilic capillaries, which prevent nonspecific surface adsorption.
We first quantified the TPO:c-MPL binding affinity in the absence of IFN-γ by titration of fluorescently labeled TPO with a soluble fragment of the c-MPL extracellular domain (residues Ser24-Trp491) as a biological proxy. In agreement with a previous study,34 TPO and c-MPL displayed a specific 2-site interaction with dissociation constants (KD) of 0.15 ± 0.04 nM and 1066 ± 126 nM, characterizing a high- and low-affinity binding site, respectively (Figure 3A; Table 1). The high-affinity interaction involves the initial contact between TPO and 1 c-MPL subunit; the low-affinity site engages the second c-MPL subunit into the complex to form a signaling-competent receptor conformation. Radiolabeled ligand assays on CD34+ cells using 125I-TPO validated the binding-affinity constants determined by MST (supplemental Figure 5D-E).
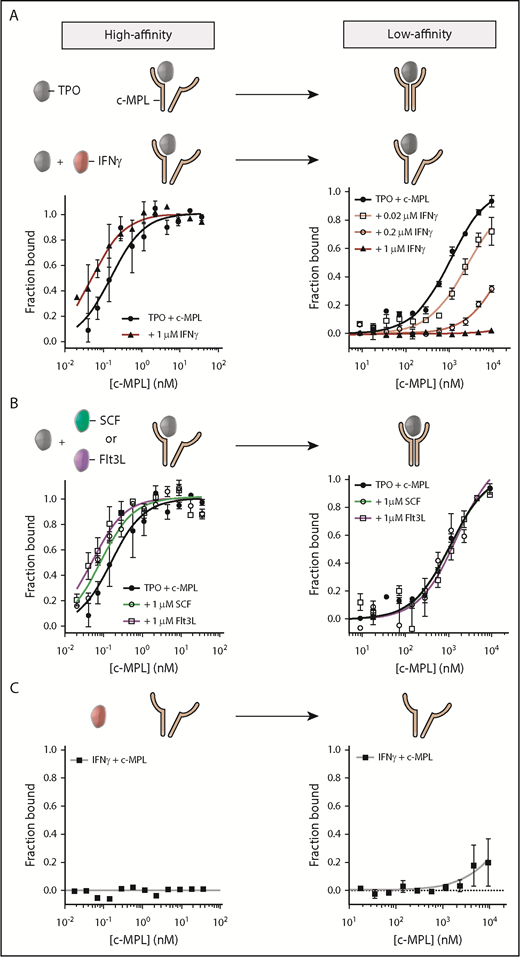
IFN-γ disrupts the low-affinity binding interaction between TPO and c-MPL. (A) Evaluation of the TPO:c-MPL binding affinity by MST in the presence (red) or absence (black) of IFN-γ. Without IFN-γ, the TPO:c-MPL interaction was characterized by a high-affinity (left panel) and a low-affinity (right panel) binding site. Addition of IFN-γ resulted in specific disruption of the low-affinity site in a dose-dependent manner. (B) Evaluation of the TPO:c-MPL binding affinity by MST in the presence of 100-fold molar excess concentration of hematopoietic cytokines (SCF and Flt3L). The TPO:c-MPL interaction was unaffected by SCF or Flt3L. (C) Evaluation of the binding affinity between IFN-γ and c-MPL by MST. IFN-γ did not interact with c-MPL. Data are mean ± SEM; curve fit by nonlinear regression. The KD for each condition is listed in Table 1.
IFN-γ disrupts the low-affinity binding interaction between TPO and c-MPL. (A) Evaluation of the TPO:c-MPL binding affinity by MST in the presence (red) or absence (black) of IFN-γ. Without IFN-γ, the TPO:c-MPL interaction was characterized by a high-affinity (left panel) and a low-affinity (right panel) binding site. Addition of IFN-γ resulted in specific disruption of the low-affinity site in a dose-dependent manner. (B) Evaluation of the TPO:c-MPL binding affinity by MST in the presence of 100-fold molar excess concentration of hematopoietic cytokines (SCF and Flt3L). The TPO:c-MPL interaction was unaffected by SCF or Flt3L. (C) Evaluation of the binding affinity between IFN-γ and c-MPL by MST. IFN-γ did not interact with c-MPL. Data are mean ± SEM; curve fit by nonlinear regression. The KD for each condition is listed in Table 1.
High- and low-affinity binding constants of TPO:c-MPL in the presence or absence of cytokines
| Proteins | High-affinity KD, nM | Low-affinity KD, nM |
|---|---|---|
| TPO:c-MPL | 0.15 ± 0.04 | 1 066 ± 126 |
| TPO:c-MPL + 1 µM IFN-γ | 0.05 ± 0.01 | 3.72 × 1018* |
| TPO:c-MPL + 0.2 µM IFN-γ | n.d. | 23 592 ± 19 977 |
| TPO:c-MPL + 0.02 µM IFN-γ | n.d. | 2 394 ± 593 |
| TPO:c-MPL + 1 µM SCF | 0.08 ± 0.01 | 1 118 ± 412 |
| TPO:c-MPL + 1 µM Flt3L | 0.05 ± 0.01 | 1 556 ± 582 |
| Proteins | High-affinity KD, nM | Low-affinity KD, nM |
|---|---|---|
| TPO:c-MPL | 0.15 ± 0.04 | 1 066 ± 126 |
| TPO:c-MPL + 1 µM IFN-γ | 0.05 ± 0.01 | 3.72 × 1018* |
| TPO:c-MPL + 0.2 µM IFN-γ | n.d. | 23 592 ± 19 977 |
| TPO:c-MPL + 0.02 µM IFN-γ | n.d. | 2 394 ± 593 |
| TPO:c-MPL + 1 µM SCF | 0.08 ± 0.01 | 1 118 ± 412 |
| TPO:c-MPL + 1 µM Flt3L | 0.05 ± 0.01 | 1 556 ± 582 |
Data are mean ± SEM.
n.d., not done.
Error is not reported due to the ambiguity of the nonlinear regression.
To investigate the impact of IFN-γ on each binding site of the TPO:c-MPL interaction, we titrated c-MPL into a premixed solution containing TPO and a 100-fold molar excess concentration of IFN-γ to mimic conditions of inflammation. Addition of IFN-γ did not significantly perturb the TPO:c-MPL high-affinity binding site, as indicated by a KD (0.05 ± 0.01 nM) nearly unchanged relative to the one measured in the absence of IFN-γ (0.15 ± 0.04 nM) (Figure 3A; Table 1). In contrast, the same molar excess concentration of IFN-γ completely disrupted the low-affinity binding site of the TPO:c-MPL complex (KD, not detectable) (Figure 3A; Table 1). Disruption of the TPO:c-MPL low-affinity binding site by IFN-γ was dose dependent, suggesting competition between c-MPL and IFN-γ for TPO (Figure 3A; Table 1). To confirm that this effect was specific to IFN-γ, we also challenged the TPO:c-MPL interaction with saturating amounts of other cytokines important in HSPC regulation, including SCF and Flt3L. SCF and Flt3L had no detectable impact on either binding site of the TPO:c-MPL interaction (Figure 3B; Table 1). We speculated that IFN-γ might directly and competitively bind to c-MPL, but it showed no such binding affinity (Figure 3C). From these data, we infer that IFN-γ induced a dose-dependent and specific disruption of the TPO:c-MPL low-affinity binding site without directly interacting with c-MPL.
Impact of IFN-γ on TPO- and eltrombopag-induced c-MPL internalization in human HSPCs
Because recombinant forms of the TM domain of c-MPL are unavailable, MST studies are impractical to evaluate the impact of IFN-γ on the eltrombopag:c-MPL interaction. To address this question and to provide corroborative evidence of the negative effect of IFN-γ on TPO binding to c-MPL shown by MST, we compared the kinetics of c-MPL internalization induced by TPO and eltrombopag binding, in the presence or absence of IFN-γ. We used multispectral imaging flow cytometry analysis of CD34+ HSPCs labeled with fluorophore-conjugated anti–c-MPL and anti-CD34 antibodies prior to stimulation with cytokines. The subcellular localization of c-MPL in ≥10 000 CD34+ cells was monitored at 5, 15, 30, 45, and 60 minutes after the addition of cytokines.
In the absence of IFN-γ, c-MPL internalization mediated by TPO was maximal at 30 minutes (Figure 4A). When CD34+ cells were incubated with both TPO and IFN-γ, we observed a maximal reduction in TPO-mediated c-MPL internalization at 30 minutes, with a delay in peak internalization to 45 minutes, relative to conditions without IFN-γ (Figure 4A). Consistent with the observation that IFN-γ does not directly interact with c-MPL (Figure 3C), IFN-γ alone did not significantly alter internalization of c-MPL (Figure 4A), suggesting that the inhibitory effect of IFN-γ on this process is indirect and is not due to decreased c-MPL receptor availability. Eltrombopag-treated cells showed a fundamentally different pattern of c-MPL internalization, with a lower potency and more prolonged course of internalization over 60 minutes (Figure 4B), similar to its JAK-STAT5 activation profile (Figure 2D). Addition of IFN-γ to eltrombopag led to only a minimal decrease in c-MPL internalization 30 and 60 minutes after HSPC stimulation, with no statistically significant differences at 45 minutes (Figure 4B). Accordingly, the fold decrease in internalization scores caused by IFN-γ at 30, 45, and 60 minutes was significantly greater for TPO-stimulated cells than for eltrombopag-stimulated cells (Figure 4C). Thus, these data further support that IFN-γ negatively impacts human HSPCs by altering properties of the TPO:c-MPL interaction, whereas eltrombopag partly evades the interference by IFN-γ.
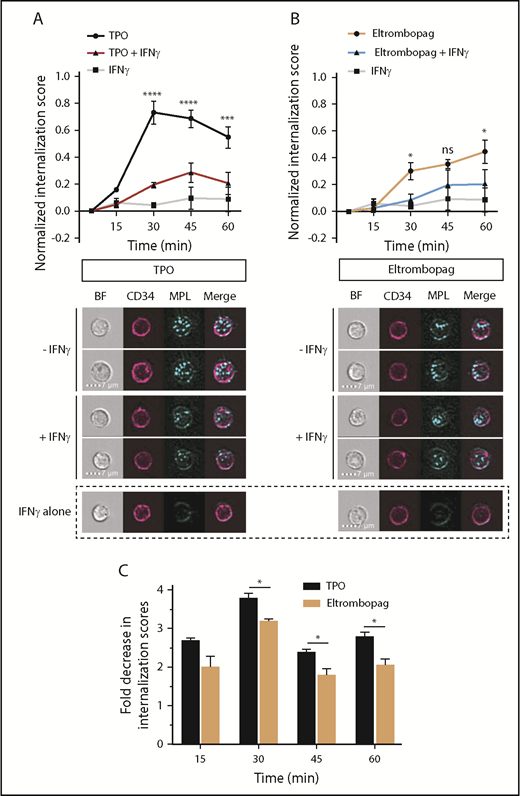
Impact of IFN-γ on TPO- and eltrombopag-induced c-MPL internalization in human HSPCs. (A) Time course of c-MPL internalization upon exposure of human CD34+ cells to TPO, with or without IFN-γ, or to IFN-γ alone. P values shown were calculated by comparing TPO with TPO + IFN-γ (upper panel). Representative images of CD34+ cells 30 minutes after exposure to TPO, TPO + IFN-γ, or IFN-γ alone (lower panel). (B) Time course of c-MPL internalization upon exposure of human CD34+ cells to eltrombopag with or without IFN-γ, or to IFN-γ alone. P values shown were calculated by comparing eltrombopag with eltrombopag + IFN-γ (upper panel). Representative images of CD34+ cells 30 minutes after exposure to eltrombopag, eltrombopag + IFN-γ, or IFN-γ alone (lower panel). (C) Quantification of the fold decrease in internalization scores caused by exposure to IFN-γ for CD34+ cells cultured with TPO or eltrombopag as a function of time. Internalization scores were calculated as described. Internalization data were obtained using multispectral imaging flow cytometry. Data are mean ± SEM. *P < .05, ***P < .001, ****P < .0001, repeated measures 2-way ANOVA with Sidak’s multiple-comparison test or multiple 2-tailed unpaired Student t tests (n = 3). BF, brightfield; MPL, c-MPL receptor.
Impact of IFN-γ on TPO- and eltrombopag-induced c-MPL internalization in human HSPCs. (A) Time course of c-MPL internalization upon exposure of human CD34+ cells to TPO, with or without IFN-γ, or to IFN-γ alone. P values shown were calculated by comparing TPO with TPO + IFN-γ (upper panel). Representative images of CD34+ cells 30 minutes after exposure to TPO, TPO + IFN-γ, or IFN-γ alone (lower panel). (B) Time course of c-MPL internalization upon exposure of human CD34+ cells to eltrombopag with or without IFN-γ, or to IFN-γ alone. P values shown were calculated by comparing eltrombopag with eltrombopag + IFN-γ (upper panel). Representative images of CD34+ cells 30 minutes after exposure to eltrombopag, eltrombopag + IFN-γ, or IFN-γ alone (lower panel). (C) Quantification of the fold decrease in internalization scores caused by exposure to IFN-γ for CD34+ cells cultured with TPO or eltrombopag as a function of time. Internalization scores were calculated as described. Internalization data were obtained using multispectral imaging flow cytometry. Data are mean ± SEM. *P < .05, ***P < .001, ****P < .0001, repeated measures 2-way ANOVA with Sidak’s multiple-comparison test or multiple 2-tailed unpaired Student t tests (n = 3). BF, brightfield; MPL, c-MPL receptor.
Formation of heteromers between TPO and IFN-γ
We next investigated the mechanism by which IFN-γ disrupted the low-affinity binding interaction between TPO and c-MPL. Because IFN-γ induced a dose-dependent disruption (Figure 3A) without directly interacting with c-MPL (Figure 3C), we hypothesized that IFN-γ may instead bind to TPO, forming heteromeric complexes that sterically hinder the low-affinity TPO:c-MPL binding site, whereas the nonpeptidyl small molecule eltrombopag could evade that process.
We tested the possibility of TPO:IFN-γ heteromer formation using MST. Notably, these experiments revealed a specific 1-site heteromeric interaction between TPO and fluorescently labeled IFN-γ (Figure 5A), with an affinity (KD = 488 ± 76 nM; Table 2) superior to the TPO:c-MPL low-affinity interaction (KD = 1066 ± 126 nM; Table 1). By contrast, no interaction was observed between IFN-γ and other early-acting hematopoietic cytokines (SCF and Flt3L) (Figure 5A; Table 2). When TPO was used as the labeled binding partner, we detected a similar high-affinity TPO:IFN-γ interaction (KD = 402 ± 89 nM) and no evidence of complex formation between TPO and SCF or Flt3L (Figure 5B; Table 2). Eltrombopag had no affinity for IFN-γ in the nanomolar to high micromolar concentration range, as demonstrated by MST (Figure 5C) and fluorescence spectroscopy (Figure 5D). Thus, formation of TPO:IFN-γ heteromers may represent a molecular mechanism by which IFN-γ hinders TPO:c-MPL interaction in human HSPCs. Moreover, because eltrombopag cannot complex with IFN-γ, this finding provides a rational explanation for its ability to overcome IFN-γ–mediated obstruction of TPO cell signaling pathways (Figure 5E).
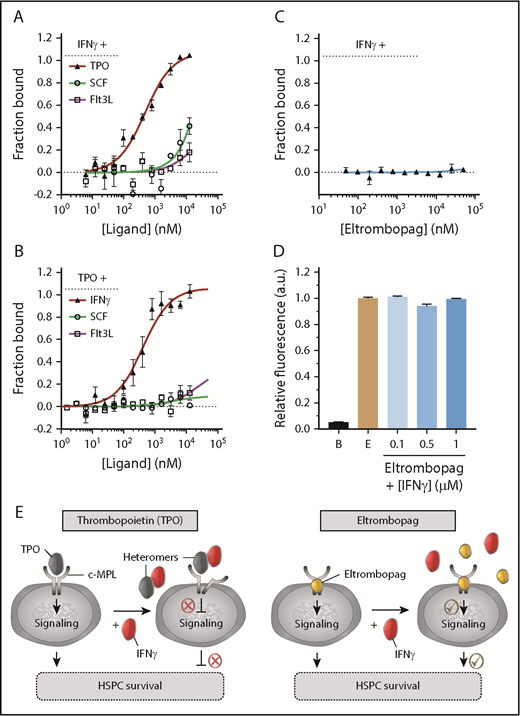
Specific high-affinity interaction between TPO and IFN-γ. (A) Evaluation of the binding affinity between NT647-labeled IFN-γ and TPO, SCF, or Flt3L using MST. Specific IFN-γ:TPO heteromer formation was detected. (B) Evaluation of the binding affinity between NT647-labeled TPO and IFN-γ, SCF, or Flt3L using MST. Specific TPO:IFN-γ heteromer formation was detected. (C) Evaluation of the binding affinity between eltrombopag and IFN-γ in the presence of 1000-fold molar excess concentration of eltrombopag using MST. IFN-γ did not interact with eltrombopag. (D) Fluorescence far-red emission of eltrombopag (E; 665-720 nm) upon excitation at 470 nm showed no changes in intensity, even at saturating concentrations of IFN-γ, indicating no interaction between IFN-γ and eltrombopag. Background fluorescence of buffer alone (B) is shown. (E) Model depicting TPO:IFN-γ heteromers impairing TPO:c-MPL interaction, downstream cell signaling, and survival of human HSPCs (left panel). Eltrombopag does not complex with IFN-γ and can overcome the IFN-γ–mediated obstruction of TPO cell signaling pathways, resulting in improved HSPC survival (right panel). (A-B) KD values are listed in Table 2. (A-D) Data are mean ± SEM; curve fit by Langmuir isotherm nonlinear regression.
Specific high-affinity interaction between TPO and IFN-γ. (A) Evaluation of the binding affinity between NT647-labeled IFN-γ and TPO, SCF, or Flt3L using MST. Specific IFN-γ:TPO heteromer formation was detected. (B) Evaluation of the binding affinity between NT647-labeled TPO and IFN-γ, SCF, or Flt3L using MST. Specific TPO:IFN-γ heteromer formation was detected. (C) Evaluation of the binding affinity between eltrombopag and IFN-γ in the presence of 1000-fold molar excess concentration of eltrombopag using MST. IFN-γ did not interact with eltrombopag. (D) Fluorescence far-red emission of eltrombopag (E; 665-720 nm) upon excitation at 470 nm showed no changes in intensity, even at saturating concentrations of IFN-γ, indicating no interaction between IFN-γ and eltrombopag. Background fluorescence of buffer alone (B) is shown. (E) Model depicting TPO:IFN-γ heteromers impairing TPO:c-MPL interaction, downstream cell signaling, and survival of human HSPCs (left panel). Eltrombopag does not complex with IFN-γ and can overcome the IFN-γ–mediated obstruction of TPO cell signaling pathways, resulting in improved HSPC survival (right panel). (A-B) KD values are listed in Table 2. (A-D) Data are mean ± SEM; curve fit by Langmuir isotherm nonlinear regression.
Binding affinities of IFN-γ–NT647 or TPO-NT647 with cytokines
| Proteins | KD (nM) |
|---|---|
| IFN-γ–NT647:TPO | 487.5 ± 76.1 |
| IFN-γ–NT647:SCF* | 3.08 × 1017 |
| IFN-γ–NT647:Flt3L | 24 925 ± 74 862 |
| TPO-NT647:IFN-γ | 402.1 ± 89.4 |
| TPO-NT647:SCF | 2 970 ± 6 234 |
| TPO-NT647:Flt3L | 23 037 ± 66 577 |
| Proteins | KD (nM) |
|---|---|
| IFN-γ–NT647:TPO | 487.5 ± 76.1 |
| IFN-γ–NT647:SCF* | 3.08 × 1017 |
| IFN-γ–NT647:Flt3L | 24 925 ± 74 862 |
| TPO-NT647:IFN-γ | 402.1 ± 89.4 |
| TPO-NT647:SCF | 2 970 ± 6 234 |
| TPO-NT647:Flt3L | 23 037 ± 66 577 |
Data are mean ± SEM.
Error is not reported due to the ambiguity of the nonlinear regression.
Discussion
This work stemmed from the clinical observation that eltrombopag could improve trilineage hematopoiesis in the setting of chronic inflammation in subjects with severe AA, despite already elevated levels of TPO. By directly comparing the effect of IFN-γ on human HSPCs in the presence of TPO or eltrombopag in vitro, we made 2 observations. First, we found that IFN-γ perturbed TPO-induced signaling pathways in human HSPCs and that eltrombopag could bypass this inhibition, resulting in enhanced progenitor activity and long-term HSPC repopulating potential in the presence of IFN-γ. Second, we showed that IFN-γ disrupted the low-affinity interaction between TPO and the extracellular domain of c-MPL, delineating a molecular mechanism by which IFN-γ inhibits TPO signaling in HSPCs. The fact that eltrombopag binds to an alternative site on c-MPL, within the TM domain of the receptor, could explain its ability to bypass this inhibition, despite equivalent SOCS upregulation in HSPCs stimulated with TPO and eltrombopag.
Further investigation is needed to fully understand the mechanism by which IFN-γ disrupted the low-affinity binding interaction between TPO and c-MPL in our study. The intriguing possibility of heteromeric complexation between TPO and IFN-γ will require direct biochemical evidence in the form of a crystal structure of the complex for definitive in vitro validation. Nevertheless, biophysical data presented in this study and other corroborative evidence strongly suggest a unique and specific interaction between TPO and IFN-γ. It is unlikely that the TPO:IFN-γ complex forms as a result of the cytokine respective net charges favoring nonspecific attractive interactions between these proteins. Indeed, the recombinant TPO used in our study, despite its high aliphatic index and a theoretical net-positive charge based on its elevated isoelectric point, showed a specific interaction only with IFN-γ, which is also positively charged at the pH used in our assays; there were no interactions observed with SCF or Flt3L notwithstanding their expected net-negative charges. In addition, because TPO and IFN-γ used in our study were recombinantly expressed in Escherichia coli, it is improbable that posttranslational modifications, generally observed with eukaryotic expression systems, would facilitate protein binding. Thus, these observations indicate that the TPO:IFN-γ interaction is most likely mediated by a combination of aliphatic and electrostatic interactions between specific as-yet-unidentified amino acid residues.
The TPO:IFN-γ heteromeric complexation observed by MST assays in this study will also require clinical validation. Based on the determined complex KD, it is improbable that TPO:IFN-γ heteromers would be detected in the circulation of AA patients, because plasma levels of IFN-γ are generally not elevated.42 Instead, elevated local concentrations of IFN-γ within the BM microenvironment, where most HSPCs reside, appear to be required to mediate potent hematopoietic inhibition4,47 ; thus, an inflammatory BM niche could provide an ideal milieu for the formation of such heteromers and disruption of the TPO:c-MPL interaction. In contrast, eltrombopag may bypass the inflammatory inhibition of signal transduction by avoiding capture by IFN-γ in the BM, thus providing an additional explanation for its clinical efficacy in severe AA, despite already elevated levels of TPO in these patients.
Other molecular mechanisms may also contribute to the positive impact of eltrombopag on HSPCs in AA patients. Eltrombopag was recently described as a powerful iron chelator with intracellular iron-mobilizing properties leading to decrements in iron-induced reactive oxygen species in cell culture models.48,49 Chelation was also observed in vivo in AA patients treated with eltrombopag.50 This effect has been proposed to account, in part, for the improved function of cells cultured with eltrombopag, its antiproliferative properties in acute myeloid leukemia48,51 and hepatocellular carcinoma,52 and the clinical hematopoietic benefits in subjects with AA38-40 and myelodysplastic syndrome.53 The iron-mobilizing effects were observed in human and rodent cells. Because eltrombopag activates c-MPL by association with a specific amino acid found only in humans and chimpanzees, but not in rodents,45 it has been postulated that the iron-binding properties of eltrombopag are independent of c-MPL, perhaps via direct diffusion into cells.49,54 Although data from our study confirm the established dependence of eltrombopag on c-MPL expression in HSPCs and activation of downstream signaling for its activity in target cells, they do not exclude that some of the beneficial effects of eltrombopag may be independent of c-MPL stimulation and restoration of signal transduction cascades in HSPCs. Thus, both mechanistic models are likely complementary rather than exclusionary.
In addition to the molecular mechanism underpinning the pathogenesis of AA uncovered in this study, our data provide new insights into cellular signaling pathways in HSPCs. We observed a fundamentally distinct pattern of signaling activation in CD34+ cells between TPO and eltrombopag. STAT5 phosphorylation was rapid, robust, and briskly declined with TPO, whereas eltrombopag produced a much slower and weaker signal that persisted longer. This prolonged activation of STAT5 with eltrombopag is consistent with the observed slower c-MPL internalization believed to halt signal transduction. How prolonged STAT5 signal delivery in the presence of eltrombopag in HSPCs ultimately influences cell fate will require further investigation, but some evidence shows that the magnitude, as well the persistence, of the cytokine signal is important in regulation.55 The apparent preference of eltrombopag for activation of the JAK-STAT3 pathway in our study is also important. Consistent with the reduced cell death observed with eltrombopag relative to TPO in the presence of IFN-γ, several antiapoptotic genes contain STAT3-response elements.56 In addition, STAT3 was shown to be an important regulator of hematopoietic regeneration under stimulated, but not homeostatic, conditions.57 Indeed, downregulation of STAT3 in HSPCs reduced their competitive reconstitutive ability in a murine transplant model. In contrast, upregulation of STAT3 activity markedly increased HSPC regenerative activity in lethally irradiated recipients without evidence of dysregulated hematopoiesis long-term. Thus, it is conceivable that, under inflammatory stimulation, restoration of STAT3 signaling in HSPCs by eltrombopag could similarly promote their survival and regenerative activity.
Our results have considerable implications for immune-mediated BM failure, as well as for disorders of chronic inflammation in general. In addition to AA, eltrombopag might ameliorate the pancytopenia accompanying other disorders characterized by inflammation and marrow failure, such as chronic infections, as well as posttransplant graft-versus-host disease or trilineage graft failure. Published case reports support the use of eltrombopag after allogeneic HSPC transplantation58-62 ; prospective clinical trials will further inform on this possible therapeutic application. Moreover, analogous to the negative impact of IFN-γ on TPO regulation of HSPCs in immune-mediated BM failure, suppression of erythropoiesis by inflammation is well known in the clinic. In patients with anemia of chronic inflammation, IFN-γ impairs the responsiveness of erythroid progenitor cells to EPO, the primary regulator of red blood cell production.63 Consistent with the significant sequence and structural homology between TPO and EPO at or near the receptor binding sites, as well as between their respective cognate receptors,64-70 we also observed impaired binding of EPO to the EPO receptor in the presence of IFN-γ, with subsequent interference with signaling cascades in erythroid progenitors (H.D.H., L.J.A., and A.L., manuscript in preparation). Future studies will provide the opportunity to investigate the contribution of these concepts to other pathological conditions and test novel therapeutic strategies that could stem from this new understanding.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all members of the Larochelle Laboratory for helpful discussions and particularly Richard Smith for critically reviewing the manuscript; David Stroncek and the National Institutes of Health Department of Transfusion Medicine and Cell Processing Section staff for apheresis, selection, and cryopreservation of human CD34+ cells; Kinneret Broder, Therese Intrater, Debbie Draper, Tatyana Worthy, and the outpatient clinic nursing staff for recruiting normal volunteers and providing administration teaching to healthy subjects; Crystal Thomas, James Hawkins, and the mouse core facility staff for excellent animal care; Keyvan Keyvanfar for technical support with flow cytometry; J. Philip McCoy, Venina Dominical, Leigh Samsel, and the National Heart, Lung, and Blood Institute (NHLBI) Flow Cytometry Core Facility staff for their contribution to multispectral imaging flow cytometry; Grzegorz Piszczek and the NHLBI Biophysics Core Facility staff for assistance with MST; Xin Chen for assistance with biostatistical analyses; and Rolf Swenson, Ana Opina, and Haitao Wu from the NHLBI Imaging Probe Development Center for assistance with radiolabeling of recombinant TPO.
This work was supported by the intramural research program of the National Institutes of Health, NHLBI. GlaxoSmithKline and Novartis, the manufacturers of eltrombopag, provided research-grade drug and research funding to the NHLBI under a Cooperative Research and Development Agreement. H.C. was supported by a fellowship (#81500097) from the Natural Science Foundation of China.
Authorship
Contribution: Conceptualization of the project was done by A.L., N.S.Y., C.E.D., D.M.T., T.W., and X.F.; experiments were done by L.J.A., H.D.H., H.C., and A.L.; data curation was performed by L.J.A., H.D.H., H.C., and A.L.; formal analysis of data was conducted by L.J.A., H.D.H., H.C., and A.L.; data validation was performed by H.D.H.; project was supervised by A.L., N.S.Y., and C.E.D.; original draft was written by A.L., L.J.A., and H.D.H.; review and editing of the manuscripts were performed by L.J.A., H.D.H., H.C., N.S.Y., C.E.D., D.M.T., T.W., X.F., and A.L.; funding acquisition was provided by A.L. and N.S.Y.; and project administration was done by A.L.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for L.J.A. is 10X Genomics, Inc., Pleasanton, CA.
The current affiliation for H.C. is Department of Hematology, Affiliated Hospital of Xuzhou Medical University, and Xuzhou Medical University, Xuzhou, China.
Correspondence: Andre Larochelle, National Heart, Lung, and Blood Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892; e-mail: larochea@nhlbi.nih.gov.
