

In this issue of Blood, present intriguing data that the cell surface marker NCAM1 (neural cell adhesion molecule 1, also known as CD56), which is expressed in approximately 15% to 20% of acute myeloid leukemia (AML) cases, promotes chemotherapy resistance and leukemia stem cell (LSC, also referred to as a leukemia initiating cell) function, in part through activation of the MAPK signaling cascade (see figure). Notably, they provide data that pharmacologic inhibitors of the MAPK pathway synergistically cooperate with cytarabine (Ara-C) in eliminating NCAM1-expressing AML cells, suggesting that NCAM1 may represent both a potential targetable vulnerability and a biomarker for guiding treatment decisions in AML.1-4
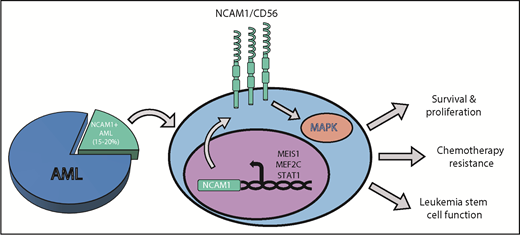
NCAM1/CD56 supports chemotherapy resistance and leukemia cell function in AML. The expression of the membrane surface protein NCAM1 or CD56 (NCAM1/CD56) is elevated in approximately 15% to 20% of AML cases. NCAM1 transcription is regulated by a series of transcription factors such as MEIS1, MEF2C, or STAT1. Once expressed and activated on the surface of AML cells, NCAM1 activates the MAPK (MAP-kinase) signaling cascade to promote cell survival and proliferation, resistance to conventional AML chemotherapies, and leukemia stem cell function.
NCAM1/CD56 supports chemotherapy resistance and leukemia cell function in AML. The expression of the membrane surface protein NCAM1 or CD56 (NCAM1/CD56) is elevated in approximately 15% to 20% of AML cases. NCAM1 transcription is regulated by a series of transcription factors such as MEIS1, MEF2C, or STAT1. Once expressed and activated on the surface of AML cells, NCAM1 activates the MAPK (MAP-kinase) signaling cascade to promote cell survival and proliferation, resistance to conventional AML chemotherapies, and leukemia stem cell function.
AML arises from the clonal outgrowth of mutated hematopoietic stem and progenitor cells that display uncharacteristic properties such as self-renewal, augmented growth, and an inability to differentiate into mature progeny.5,6 The need for more-effective therapies is clear, as many patients are refractory to first-line chemotherapies, and chemotherapy-resistant relapse is a major contributor to treatment failure. However, AML is a complex and dynamic malignancy that can arise from myriad combinations of infrequent genetic mutations in which patients frequently display multiple, mutationally diverse coexisting clones.6,7 Although efforts to target certain, more commonly mutated genes (such as FLT3 or IDH2) have shown clinical efficacy, strategies targeting nonmutated molecules or pathways that are aberrantly expressed/activated irrespective of genetic subtype are highly desirable.
NCAM1 expression is largely associated with healthy neural tissue and lymphocytic lineage cells such as NK cells, but has also been observed in numerous solid cancers and hematologic tumors. In AML, its expression is found in 15% to 20% of patients analyzed, particularly in t(8;21) and acute promyelocytic leukemia, where it identifies a subgroup of patients with a more unfavorable prognosis, extramedullary leukemia, and shorter remissions.2-4 However, before the study put forth in this issue by Sasca et al, the molecular mechanisms underlying these associations were uninvestigated.
Sasca et al show that NCAM1 expression is heterogeneous across different genetic subtypes of AML, with the exception of complex-karyotype AML (CK-AML) as well as AMLs bearing 11q23 rearrangements or t(8;21), which display significantly higher NCAM1 levels relative to other genetic subtypes.1 Sasca et al also present data that NCAM1 expression is driven by several transcription factors, such as MEIS1, MEF2C, and STAT1, but do so in a cell context manner (see figure).1
To investigate the functional role of NCAM1 in human AML cell biology, Sasca et al employ an inducible shRNA system and observe that inhibition of NCAM1 expression selectively inhibits the growth of NCAM1-positive (NCAM1+) human AML cells, both in vitro and in vivo. Notably, Sasca et al also show that NCAM1 inhibition renders NCAM1+ AML cells more sensitive to the first-line AML chemotherapy Ara-C in vivo, and that enforced NCAM1 expression partially protects NCAM1-negative (NCAM1−) AML cells from Ara-C or daunorubicin treatment in vitro.
Given that LSCs are purported to be the architects of disease relapse and chemotherapy evasion,8 Sasca et al explored the role of NCAM1 in LSC biology in a mouse model of AML driven by the 11q23 rearrangement, MLL-AF9. A comparison of various malignant and healthy murine hematopoietic stem and progenitor cell populations revealed that MLL-AF9-expressing LSCs display significantly higher levels of Ncam1 compared with all other populations. Importantly, Sasca et al also show that deletion of Ncam1 significantly extended the time of disease onset and drastically reduced the frequency of LSCs in this model.
Using a combination of phospho-proteomic and RNA-seq analyses, Sasca et al found that inhibition of NCAM1 diminished several signal transduction pathways, including MAPK signaling, as well as transcriptional programs related to glucose metabolism and hypoxia. Capitalizing on these observations, they also show evidence that NCAM1+ AML cells are significantly more sensitive to MAPK inhibitors compared with NCAM1− AML cell lines, and that the MAPK inhibitor trametinib synergistically cooperates with Ara-C to eliminate NCAM1+ AML cell lines.
The data presented by Sasca et al have several potential clinically relevant ramifications. First, the presented data that the NCAM1 pathway supports leukemia cell survival makes it an appealing therapeutic target, particularly for NCAM1-expressing CK-AML and 11q23 rearranged AMLs, which are notoriously associated with poor response to current therapies. However, given that NCAM1 is widely expressed in the nervous system and other hematopoietic populations, it will be essential for future studies to focus on identifying and targeting the downstream molecular events that NCAM1 engages to uniquely support AML. Second, NCAM1 may represent a biomarker for guiding treatment decisions. For example, clinical trials evaluating MAPK inhibitors in AML have primarily focused on patients expressing RAS mutations; however, these studies have yielded limited to moderate efficacy.9,10 The results presented by Sasca et al suggest that NCAM1 may be a putative biomarker for identifying patients that might benefit from MAPK inhibitor-based therapies, particularly in combination with Ara-C. Third, the observations that NCAM1 marked and functionally supported LSCs in a mouse model of AML may be relevant in NCAM1+ human AML. For example, if NCAM1 is expressed on LSC/preleukemia clone populations in human AML, NCAM1 may serve as a marker for identifying or possibly therapeutically targeting such populations. Therefore, it will be important in the future to assess NCAM1 expression across coexisting leukemia and preleukemia clones within a given patient sample to fully understand the clinical potential of this surface protein in AML.
Conflict-of-interest disclosure: The author declares no competing financial interests.

