

Key Points
Aberrant NCAM1 expression confers drug resistance in AML.
Expression of NCAM1 activates the MAPK–extracellular signal-regulated kinase pathway and renders cells sensitive to MEK inhibitors.

Abstract
Neural cell adhesion molecule 1 (NCAM1; CD56) is expressed in up to 20% of acute myeloid leukemia (AML) patients. NCAM1 is widely used as a marker of minimal residual disease; however, the biological function of NCAM1 in AML remains elusive. In this study, we investigated the impact of NCAM1 expression on leukemogenesis, drug resistance, and its role as a biomarker to guide therapy. Beside t(8;21) leukemia, NCAM1 expression was found in most molecular AML subgroups at highly heterogeneous expression levels. Using complementary genetic strategies, we demonstrated an essential role of NCAM1 in the regulation of cell survival and stress resistance. Perturbation of NCAM1 induced cell death or differentiation and sensitized leukemic blasts toward genotoxic agents in vitro and in vivo. Furthermore, Ncam1 was highly expressed in leukemic progenitor cells in a murine leukemia model, and genetic depletion of Ncam1 prolonged disease latency and significantly reduced leukemia-initiating cells upon serial transplantation. To further analyze the mechanism of the NCAM1-associated phenotype, we performed phosphoproteomics and transcriptomics in different AML cell lines. NCAM1 expression strongly associated with constitutive activation of the MAPK-signaling pathway, regulation of apoptosis, or glycolysis. Pharmacological inhibition of MEK1/2 specifically inhibited proliferation and sensitized NCAM1+ AML cells to chemotherapy. In summary, our data demonstrate that aberrant expression of NCAM1 is involved in the maintenance of leukemic stem cells and confers stress resistance, likely due to activation of the MAPK pathway. Targeting MEK1/2 sensitizes AML blasts to genotoxic agents, indicating a role for NCAM1 as a biomarker to guide AML treatment.
Introduction
Acute myeloid leukemia (AML) is a clonal disease of the hematopoietic stem and progenitor cell (HSPC).1 During the course of leukemogenesis, HSPCs consecutively acquire genetic alterations, which in concert rewire transcriptional networks and ultimately result in clonal growth advantage, aberrant self-renewal, and disturbed differentiation.1 Leukemic blasts are characterized by the expression of a broad spectrum of cell surface molecules called cluster of differentiation (CD).2 Expression of defined CD markers can indicate biological properties, for example, CD34 as a surrogate marker for leukemic stem cells (LSCs)3,4 ; the commitment to specific lineages, for example, CD64 as a marker for monocytic differentiation5 ; or function as a therapeutic target, for example, gemtuzumab ozogamicin directed against CD33.6 Interestingly, myeloid blasts frequently express CD markers associated with lymphoid lineages such as CD2, CD7, CD19, and CD56.7 These markers might simply represent a remnant of disturbed evolution of stem cells toward committed progenitors. Alternatively, aberrant expression of lymphoid markers could result from novel transcriptional programs evoked by defined genetic lesion, for instance, CD19 expression in mixed lineage leukemia (MLL)–rearranged leukemia.8 Lymphoid markers are particularly suitable for monitoring minimal residual disease, however, little or nothing is known about their functional relevance.9
In human hematopoiesis, CD56, also known as neural cell adhesion molecule 1 (NCAM1) is highly expressed in natural killer (NK) cells.10 In addition, NCAM1 expression was found in rare subsets of T and B lymphocytes, dendritic cells, and neural or mesenchymal stem cells.11-13 Moreover, NCAM1 is transiently, but extensively, expressed during embryogenesis.14
NCAM1 belongs to the immunoglobulin superfamily of adhesion molecules.15 A wide range of alternatively spliced NCAM1 messenger RNAs (mRNAs) has been described to date, but only the 120-, 140-, and 180- kDa isoforms are commonly expressed.16 NCAM1 plays an important role in the regulation of neurogenesis, neurite outgrowth, proliferation, and cell migration, however, its function in hematopoiesis, including NK cells, is poorly understood. NCAM1 signaling is mediated either by homophilic or heterophilic interactions with fibroblast growth factor receptor (FGFR), L1-CAM, N-cadherin and other components of the extracellular matrix.16,17 Upon activation, NCAM1 triggers a variety of signaling cascades including FYN–focal adhesion kinase (FAK), MAPK, and phosphatidylinositol 3-kinase (PI3K) pathways.18-20
NCAM1140 represents the major isoform expressed in cancer cells and has also been described in several tumor entities such as glioma, ovarian cancer, and small cell lung cancer. Functionally, NCAM1 expression is linked to disease progression.21-23 In AML, aberrant expression of NCAM1 has also been reported in ∼15% to 20% of patients and was associated with reduced complete remission rates, higher relapse rates, and poor overall survival.24,25 NCAM1 as a prognostic marker was found in several cytogenetic and molecular AML subtypes, but was most evident in AML patients harboring t(8;21) AML or acute promyelocytic leukemia.24,26 NCAM1 expression also correlated with an increased risk of extramedullary disease and hyperleukocytic syndromes.27-29 These studies indicate a functional role of NCAM1 in disease progression and resistance to therapy. In line with this, previous reports demonstrated NCAM1-dependent expression of NF-κB and BCL2.30
In this study, we comprehensively analyze the impact of aberrant NCAM1 expression on leukemogenesis and the role of NCAM1 in drug resistance and as a potential biomarker for therapeutic strategies.
Materials and methods
Cell culture
HL-60, K562, Kasumi-1, MonoMac-6, MV4-11, THP1, OCI-AML2, OCI-AML3, NOMO-1, U937, and SKM-1 cell lines were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). MOLM-14 cells were a gift from Scott Armstrong (Dana-Farber Cancer Institute, Boston Children’s Hospital, Boston, MA). AML cell lines were cultured as recommended by the DSMZ. 293FT cells were grown in Dulbecco modified Eagle medium (Gibco) and 10% fetal bovine serum (FBS) (Biochrom). Cells were expanded 1 to 3 weeks in liquid culture prior to any experiment and routinely checked for mycoplasma contamination using the Venor GeM Mycoplasma Detection kit. Cell line identity and purity were regularly verified (Multiplexion).
Patient samples
Heparin-treated bone marrow (BM) and/or leukapheresis samples were obtained from patients with AML treated at the University Medical Center (Mainz, Germany). Informed consent was obtained in accordance with the Declaration of Helsinki. All laboratory experiments with primary samples were approved by the local ethics committee.
Xenotransplantation assays
Eight-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) were transplanted with 1 × 106 of short hairpin (sh)_1-, sh_2- or scrambled (Scr)-expressing NOMO-1 cells via tail-vein injection. Doxycycline (Dox; 2 mg/mL) and 1% sucrose were added to the drinking water every 2 days between days +15 and +55. Cytosine arabinoside (Ara-C) was applied once daily intraperitoneally between days +20 and +30. Mice were euthanized at the first signs of disease. All animal studies were conducted in compliance with institutional guidelines and were approved by the responsible regulatory authorities.
Animals, retroviral transduction, and BM transplantation
Eight- to 10-week-old C57BL/6J (B6) mice were obtained from the local animal facility. Germline Ncam1 knockout (N−/−) mice were kindly provided by Harold Cremer (IBDM, Aix-Marseille Université, CNRS Marseille, France).31 All animal studies were conducted in compliance with institutional guidelines and were approved by regulatory authorities.
Mll-Af9 retroviral particles were generated as previously described.32 N−/− or B6 BM cells were transduced via spinoculation and 5 × 106 cells were transplanted into lethally irradiated (9.5 Gy) mice. Recipients of cells previously cultured in methylcellulose were cotransplanted with 2 × 105 whole BM helper cells. Secondary and tertiary transplantations were performed upon sublethal irradiation of recipients (4.75 Gy). All mice were euthanized at first signs of disease.
Statistics
Unless otherwise specified, data are presented as mean ± standard deviation (SD). Comparisons between 2 groups were performed using the unpaired Student t test. P < .05 was considered significant. For animal studies, Kaplan-Meier survival analysis was performed and survival was calculated using the log-rank test. Statistical computations were performed using GraphPad Prism software, versions 5.0 and 7.0. The number of leukemia-initiating cells was calculated using L-Calc Limiting Dilution Software (Stemcells).
Extended methods for lentiviral transduction, cell viability and proliferation assays, DNAseI hypersensitivity assays, RNA sequencing (RNA-seq)–, flow cytometry– and mass spectrometry–related preparation and analysis are available as supplemental Materials and methods (available on the Blood Web site).
Results
NCAM1 is heterogeneously expressed in all AML subsets
To gain insight into the frequency and level of NCAM1 expression, we analyzed a panel of AML cell lines (n = 11; supplemental Table 1) and primary AML patient samples (n = 16; supplemental Table 2) by flow cytometry and quantitative reverse transcription–polymerase reaction (qRT-PCR). NCAM1 expression was highly variable, ranging from absent to high levels in cell lines and primary AML blasts (Figure 1A-B). Cell surface expression of NCAM1 correlated positively with mRNA levels, suggesting transcriptional regulation of NCAM1 expression (Figure 1C). To address the question of whether expression of NCAM1 is linked to defined AML genotypes or patient characteristics, we analyzed several publicly available gene expression data sets of AML cohorts (GSE6891, GSE17855, GSE15434, TCGA).33-36 In line with previous reports, high NCAM1 expression was detected in t(8;21) leukemia24 (Figure 1D). In addition, high NCAM1 expression was significantly associated with AML samples harboring 11q23 rearrangements or complex karyotypes (Figure 1D). In the latter, no relationship with TP53 mutation status was detected. NCAM1 expression was also significantly increased in AML with mutant cKIT and RAS but this association was due to comutation with t(8;21) AML (Figure 1E; supplemental Figure 2A). Nonetheless, expression of NCAM1 was highly heterogeneous in almost all cytogenetic and molecular subgroups, ranging from absent to high levels. Finally, NCAM1 expression was detected irrespective of French-American-British (FAB) classifications or patient age but associated with incremental blast percentage in the BM (supplemental Figure B-D). These data demonstrate that NCAM1 expression is found in most subtypes of AML, which precludes any clear correlation with AML classification systems (World Health Organization [WHO]; European LeukemiaNet [ELN]).37,38
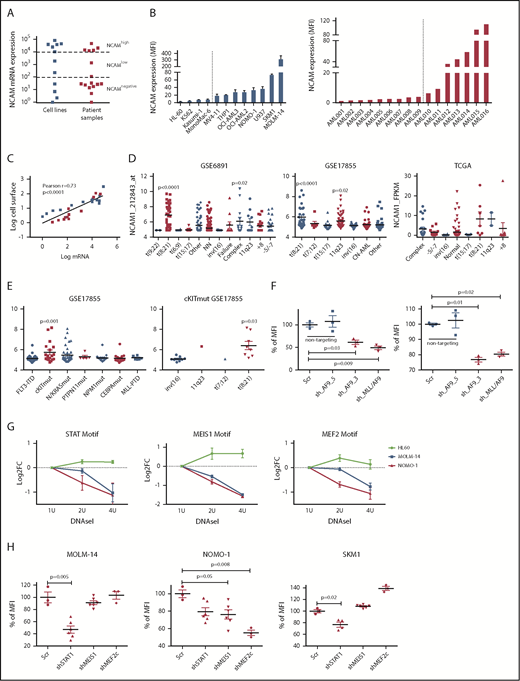
NCAM1 is heterogeneously expressed in all AML subsets. (A) Quantitative mRNA expression of NCAM1 was assessed in 11 AML cell lines (blue) and 16 patient samples (red). Cycle threshold (CT) values were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and log-fold expression is shown relative to the sample with lowest NCAM1 expression for each group. Cutoff values were arbitrarily chosen as <1 × 102 for NCAM−, 1 × 102-1 × 104 for NCAMlow, and >1 × 104 for NCAMhigh samples. (B) Cell surface expression of NCAM1 relative to isotypic immunoglobulin G (IgG) control in indicated AML cell lines (left panel) and patient samples (right panel) was analyzed by flow cytometry. Dotted lines separate NCAM1− from NCAM1+ samples. Error bars represent mean of 3 measurements ± SD of mean. (C) Correlation of NCAM1 mRNA and cell surface expression (log10 transformed values; Pearson correlation, r). (D-E) Analysis of NCAM1 expression across different cytogenetic (D) and molecular (E) groups derived from the indicated data sets. Analysis of variance (ANOVA) was significant at P < .01 in all comparisons. Shown P values represent significant differences between the corresponding group and mean values of all other groups. (F) Flow cytometry analysis of NCAM1 cell surface expression was performed 3 days after Dox-mediated induction of shRNA (shAF9_3′, shAF9_5′, shMLL-AF9 or Scr) as indicated. Shown is the mean fluorescence intensity (MFI) relative to Scr_shRNA controls and was compared by the 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (G) qPCR after DNAseI treatment at regions flanking indicated motifs near the NCAM1 transcription start site (TSS). The y-axis shows log2-fold changes (FC) between the Ct values at baseline (1 U) and increasing DNAseI concentrations (2 U and 4 U). Shown are results of 2 independent experimental replicates for each cell line. (H) Flow cytometry analysis of NCAM1 cell surface expression was performed 3 days after Dox-mediated induction of shRNA expression directed against STAT1, MEIS1, MEF2c, or Scr as indicated. Shown is the MFI relative to Scr_shRNA controls. Differences were compared using the 2-tailed Student t test. Error bars represent mean of 3 to 6 measurements ± SD of mean.
NCAM1 is heterogeneously expressed in all AML subsets. (A) Quantitative mRNA expression of NCAM1 was assessed in 11 AML cell lines (blue) and 16 patient samples (red). Cycle threshold (CT) values were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and log-fold expression is shown relative to the sample with lowest NCAM1 expression for each group. Cutoff values were arbitrarily chosen as <1 × 102 for NCAM−, 1 × 102-1 × 104 for NCAMlow, and >1 × 104 for NCAMhigh samples. (B) Cell surface expression of NCAM1 relative to isotypic immunoglobulin G (IgG) control in indicated AML cell lines (left panel) and patient samples (right panel) was analyzed by flow cytometry. Dotted lines separate NCAM1− from NCAM1+ samples. Error bars represent mean of 3 measurements ± SD of mean. (C) Correlation of NCAM1 mRNA and cell surface expression (log10 transformed values; Pearson correlation, r). (D-E) Analysis of NCAM1 expression across different cytogenetic (D) and molecular (E) groups derived from the indicated data sets. Analysis of variance (ANOVA) was significant at P < .01 in all comparisons. Shown P values represent significant differences between the corresponding group and mean values of all other groups. (F) Flow cytometry analysis of NCAM1 cell surface expression was performed 3 days after Dox-mediated induction of shRNA (shAF9_3′, shAF9_5′, shMLL-AF9 or Scr) as indicated. Shown is the mean fluorescence intensity (MFI) relative to Scr_shRNA controls and was compared by the 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (G) qPCR after DNAseI treatment at regions flanking indicated motifs near the NCAM1 transcription start site (TSS). The y-axis shows log2-fold changes (FC) between the Ct values at baseline (1 U) and increasing DNAseI concentrations (2 U and 4 U). Shown are results of 2 independent experimental replicates for each cell line. (H) Flow cytometry analysis of NCAM1 cell surface expression was performed 3 days after Dox-mediated induction of shRNA expression directed against STAT1, MEIS1, MEF2c, or Scr as indicated. Shown is the MFI relative to Scr_shRNA controls. Differences were compared using the 2-tailed Student t test. Error bars represent mean of 3 to 6 measurements ± SD of mean.
MEIS1, MEF2C, and STAT1 are putative regulators of the NCAM1 transcription
We next wanted to analyze potential regulatory mechanisms of NCAM1 expression. As NCAM1 was highly expressed in 11q23-rearranged leukemia and many of our AML cell lines harbored MLL-AF9 translocations, we took advantage of established short hairpin RNA (shRNA) clones targeting the 3′ end of AF9, the 5′ end of AF9, or the fusion sequence of MLL-AF9.39 Transduction of MOLM-14 and NOMO-1 cells with viral constructs expressing shAF9_3′ or shMLL-AF9 caused significant downregulation of MLL-AF9 whereas shAF9_5′, which targets only wild-type AF9, or scrambled shRNA did not (supplemental Figure 1E). Subsequent qPCR analysis demonstrated significant downregulation of NCAM1 in shAF9_3′ and shMLL-AF9 expressing cell lines (Figure 1F), suggesting that MLL-AF9 is an upstream regulator of NCAM1. To identify direct regulators of NCAM1, we performed DNAseI hypersensitivity assays coupled to qRT-PCR covering transcription factor (TF)-binding motifs within the NCAM1 promoter. We decided to analyze regions with motifs of known downstream targets of MLL-fusion proteins, such as Meis Homeobox 1 (MEIS1) and myocyte enhancer factor-2 (MEF2; for MEF2C), and motifs of transcription factors linked to leukemogenesis, for example, STATs. For all of these motifs, we could confirm open chromatin in MOLM-14 and NOMO-1 cells, but not in the NCAM1− cell line HL-60 (Figure 1G). To investigate whether these TFs are functionally involved in the regulation of NCAM1 expression, we performed knockdown experiments (supplemental Figure 1F) followed by NCAM1 expression analysis. In NOMO-1, knockdown of MEIS1 and MEF2C caused significant downregulation of NCAM1, whereas STAT1 was the obvious regulator in MOLM-14 and the MLL-AF9− cell line SKM-1 (Figure 1H). These data indicate that transcriptional modulation is the main regulatory mechanism of NCAM1 expression and that cell context–dependent expression of transcription factors at least partly guides NCAM1 expression.
Suppression of NCAM1 inhibits cell growth and enhances differentiation and cell death
Aberrant NCAM1 expression might represent a sole remnant of disturbed differentiation or alternatively plays an important functional role in AML. To address this question, we decided to suppress NCAM1 expression levels by RNA interference (RNAi)-mediated knockdown. Suppression of NCAM1 expression in MOLM-14 cells using 2 different shRNA clones rapidly induced apoptotic cell death whereas cell survival was not affected in scrambled controls or the NCAM1− cell line HL-60 (supplemental Figure 2A; data not shown). Withdrawal of puromycin was followed by NCAM1 reexpression and subsequent recovery of cultured cells (supplemental Figure 2A), indicating an essential role of NCAM1 in cell survival. To distinguish between acute stress mediated by viral infection and specific effects caused by suppression of NCAM1 expression, we generated several cell lines expressing 1 of 3 Dox-inducible shRNA clones directed against NCAM1 or scrambled shRNA. Treatment with Dox caused almost complete downregulation of NCAM1 cell surface expression within 2 days in MOLM-14, SKM-1, U937, NOMO-1, and THP-1 cells (supplemental Figure 2B). Knockdown of NCAM1 significantly inhibited cell growth, metabolic activity, and induced apoptotic cell death compared with scrambled controls (Figure 2A-C; supplemental Figure 2C-E). Only moderate downregulation of NCAM1 was observed in OCI-AML3 and MV4;11 cells (supplemental Figure 2B). Here, knockdown of NCAM1 inhibited cell growth but did not induce apoptosis, whereas cell cycle analysis demonstrated an arrest in the G0/G1 phase likely responsible for the previously observed diminished growth rates (Figure 2A-C; supplemental Figure 2C). No effects were noted in the NCAM1− cell lines HL-60 and K562, which suggests on-target activity of the shRNA clones. Interestingly, in NOMO-1 and SKM-1 cells, persistent suppression of NCAM1 induced morphological changes in line with myelomonocytic differentiation and expression of the differentiation marker CD11b (Figure 2D), likely preceding apoptotic cell death.
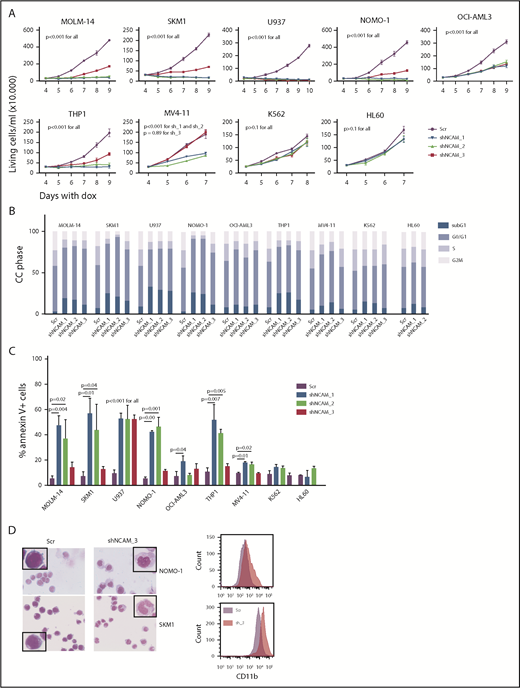
NCAM suppression inhibits cell growth and enhances differentiation and cell death. (A) Measurement of proliferation after Dox-mediated induction of different shRNA clones directed against NCAM1 (shNCAM_1, _2, _3) in the indicated cell lines. Cell count was performed daily starting at day 4 of Dox treatment. Error bars represent mean of 3 measurements ± SD of mean. (B-C) Cell cycle (CC) analysis and measurement of apoptosis were performed at day 5 after Dox-mediated induction of different shRNA clones directed against NCAM1 (shNCAM_1, _2, _3) or Scr as indicated. Shown is the fraction of cells in subG1, G1, S, and G2/M phase (B) and the percentage of Annexin V+ cells (C). Statistic analyses were performed by the 2-tailed Student t test. Error bars represent mean of 3 experiments for each condition ± SD of mean. (D) May-Grünwald-Giemsa staining and expression analysis of CD11b at day 5 after Dox-mediated induction of shNCAM_3 or Scr shRNA. Scale bar, 10 μM; the enlarged cells in the corners are 4× enhanced. Shown are representative examples of myelomonocytic differentiation and CD11b cell surface expression (N = 3).
NCAM suppression inhibits cell growth and enhances differentiation and cell death. (A) Measurement of proliferation after Dox-mediated induction of different shRNA clones directed against NCAM1 (shNCAM_1, _2, _3) in the indicated cell lines. Cell count was performed daily starting at day 4 of Dox treatment. Error bars represent mean of 3 measurements ± SD of mean. (B-C) Cell cycle (CC) analysis and measurement of apoptosis were performed at day 5 after Dox-mediated induction of different shRNA clones directed against NCAM1 (shNCAM_1, _2, _3) or Scr as indicated. Shown is the fraction of cells in subG1, G1, S, and G2/M phase (B) and the percentage of Annexin V+ cells (C). Statistic analyses were performed by the 2-tailed Student t test. Error bars represent mean of 3 experiments for each condition ± SD of mean. (D) May-Grünwald-Giemsa staining and expression analysis of CD11b at day 5 after Dox-mediated induction of shNCAM_3 or Scr shRNA. Scale bar, 10 μM; the enlarged cells in the corners are 4× enhanced. Shown are representative examples of myelomonocytic differentiation and CD11b cell surface expression (N = 3).
NCAM1 expression confers a drug-resistance phenotype
Beside its effects on cell growth, survival, and differentiation, NCAM1 might also be involved in the regulation of cellular stress response. In contrast to conditional knockdown, straight knockdown of NCAM1 induced rapid and almost complete cell death, which might result from additional stress due to simultaneous viral infection. To explore whether the loss of NCAM1 sensitizes cells to therapy, we treated several cell lines with different cytotoxic agents and analyzed cell growth and apoptosis. As shown in Figure 3A, knockdown of NCAM1 sensitized leukemic cells to Ara-C treatment and decreased the 50% inhibitory concentration (IC50) by up to 2 log10-fold. Consistent with these results, knockdown of NCAM1 significantly enhanced apoptosis upon treatment with Ara-C and daunorubicin compared with scrambled controls (supplemental Figure 3). Similar results were seen in FLT3-ITD+ MOLM-14 cells upon treatment with the FLT3-inhibitor midostaurin (Figure 3B). In contrast, exogenous overexpression of the cancer-associated21-23 splice variant NCAM1140 in the NCAM1− cell line HL-60 conferred resistance to chemotherapy and blocked all trans retinoic acid (ATRA)–induced differentiation (Figure 3C-E). Furthermore, expression of NCAM1140 in BCR-ABL+ K562 cells increased the IC50 of dasatinib from 0.97 nM to 2.2 nM compared with empty vector control cells (Figure 3F). To analyze the functional role of NCAM1 in vivo, we performed xenotransplantation experiments using an NSG mouse model. NOMO-1 cells engineered to stably express Dox-inducible shNCAM_2, shNCAM_3, or scrambled control shRNAs were transplanted and recipient mice were treated with Dox starting at day 15 posttransplantation until day 55. As shown in Figure 3G, knockdown of NCAM1 significantly increased disease latency compared with scrambled controls. Strikingly, shNCAM_2 mice survived up to 35 days after Dox treatment was stopped. Moreover, downregulation of NCAM1 expression sensitized leukemic cells to Ara-C treatment resulting in prolonged survival in shNCAM_3-transplanted animals (Figure 3G). Together, these data provide strong evidence that NCAM1 confers resistance against chemotherapy and specific kinase inhibitors.
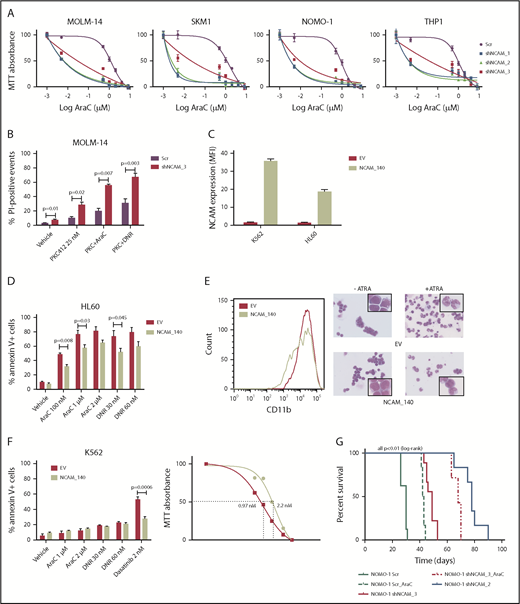
NCAM1 expression confers a drug-resistance phenotype. (A) Cell lines were exposed to Dox for 4 days and different concentrations of Ara-C (0.1- 8 µM) for 48 hours. Cell proliferation was assessed by MTT absorbance at day 4. Error bars represent mean of 3 measurements ± SD of mean. (B) MOLM-14 cells were exposed to Dox for 4 days and either DMSO (vehicle), 25 nM PKC412, Ara-C (1 μM) or daunorubicin (DNR; 30 nM) for 48 hours. Cell death was assessed by flow cytometry–based measurement of the propidium iodide–positive population and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (C-E) The NCAM1− cell lines K562 and HL60 were transduced with lentiviral particles either expressing the NCAM1_140 isoform (NCAM_140) or empty vector (EV) control. (C) Expression of NCAM1 was analyzed by flow cytometry and shown as MFI values relative to IgG isotype control. HL60 cells were exposed to DMSO (vehicle), Ara-C, or daunorubicin as indicated. (D) Apoptosis was assessed by flow cytometry–based analysis of Annexin V/7-AAD double-positive population after treatment of 24 hours as indicated and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (E) Differentiation was explored upon treatment with 2 μM ATRA or vehicle (DMSO) for 48 hours. Left, CD11b cell surface expression analysis by flow cytometry; right, images of cells stained with Max-Grünwald-Giemsa (scale bar, 10 μM; the enlarged cells in the corners are 4× enhanced). Shown are representative examples of 3 independent experiments. (F) EV- or NCAM_140-overexpressing K562 cells were treated for 48 hours as indicated. Apoptosis was assessed by flow cytometry-based analysis of Annexin V/7-AAD double-positive population and compared by 2-tailed Student t test (left). Error bars represent mean of 3 measurements ± SD of mean. Right, K562 cells were treated with different concentrations of dasatinib and inhibition of proliferation was assessed by MTT absorbance. (G) NSG mice (N = 5 per group) were engrafted with 1 × 106 NOMO-1 cells expressing either Scr, shNCAM_2, or shNCAM_3 shRNA clones. All mice received Dox starting on day 15, which was discontinued in living animals on day 55. NOMO-1_Scr and shNCAM3 were either treated with vehicle or Ara-C (10 mg/kg) once daily subcutaneous between day 20 and 30. Survival of mice is shown as Kaplan-Meier curves.
NCAM1 expression confers a drug-resistance phenotype. (A) Cell lines were exposed to Dox for 4 days and different concentrations of Ara-C (0.1- 8 µM) for 48 hours. Cell proliferation was assessed by MTT absorbance at day 4. Error bars represent mean of 3 measurements ± SD of mean. (B) MOLM-14 cells were exposed to Dox for 4 days and either DMSO (vehicle), 25 nM PKC412, Ara-C (1 μM) or daunorubicin (DNR; 30 nM) for 48 hours. Cell death was assessed by flow cytometry–based measurement of the propidium iodide–positive population and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (C-E) The NCAM1− cell lines K562 and HL60 were transduced with lentiviral particles either expressing the NCAM1_140 isoform (NCAM_140) or empty vector (EV) control. (C) Expression of NCAM1 was analyzed by flow cytometry and shown as MFI values relative to IgG isotype control. HL60 cells were exposed to DMSO (vehicle), Ara-C, or daunorubicin as indicated. (D) Apoptosis was assessed by flow cytometry–based analysis of Annexin V/7-AAD double-positive population after treatment of 24 hours as indicated and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (E) Differentiation was explored upon treatment with 2 μM ATRA or vehicle (DMSO) for 48 hours. Left, CD11b cell surface expression analysis by flow cytometry; right, images of cells stained with Max-Grünwald-Giemsa (scale bar, 10 μM; the enlarged cells in the corners are 4× enhanced). Shown are representative examples of 3 independent experiments. (F) EV- or NCAM_140-overexpressing K562 cells were treated for 48 hours as indicated. Apoptosis was assessed by flow cytometry-based analysis of Annexin V/7-AAD double-positive population and compared by 2-tailed Student t test (left). Error bars represent mean of 3 measurements ± SD of mean. Right, K562 cells were treated with different concentrations of dasatinib and inhibition of proliferation was assessed by MTT absorbance. (G) NSG mice (N = 5 per group) were engrafted with 1 × 106 NOMO-1 cells expressing either Scr, shNCAM_2, or shNCAM_3 shRNA clones. All mice received Dox starting on day 15, which was discontinued in living animals on day 55. NOMO-1_Scr and shNCAM3 were either treated with vehicle or Ara-C (10 mg/kg) once daily subcutaneous between day 20 and 30. Survival of mice is shown as Kaplan-Meier curves.
Ncam1 is highly expressed in LSCs
AML is initiated and maintained by rare LSCs. LSCs are characterized by aberrant self-renewal activity and endogenous resistance to standard chemotherapeutics, which limit efficient disease eradication.40 To investigate the role of Ncam1 in leukemogenesis, we took advantage of a well-characterized murine Mll-Af9 (hereafter M/A9) leukemia model, in which LSCs reside in the Lin−KithighSca1−CD34+FcgRII/III+ granulocyte-macrophage progenitor (L-GMP) population.41 We initially analyzed gene expression of Ncam1 in sorted L-GMPs, normal hematopoietic, and other leukemic progenitors derived from 3 different microarray datasets (GSE3722, GSE10627, GSE20377). Compared with all other populations, Ncam1 was significantly higher expressed in L-GMPs (Figure 4A). Of note, in some data sets Ncam1 expression was also observed in normal GMPs (GSE10627, GSE61468) indicating transient expression during normal maturation.42 To confirm these data, we transplanted M/A9-transduced GMPs into lethally irradiated C57BL/6 (B6) recipient mice and analyzed Ncam1 cell surface expression. Compared with normal HSCs (Lin−KithighSca1+), Ncam1 was 53-fold higher expressed in the LSC population (Figure 4B). In leukemic cells, highest expression levels were found in Lin−Kithigh cells and gradually declined in Lin−Kitmedium, Lin−Kitlow, and Lin+ cells, respectively (supplemental Figure 4). Contradictory to gene expression analyses (Figure 4A), we were not able to detect Ncam1 cell surface expression in normal GMPs (Figure 4B).
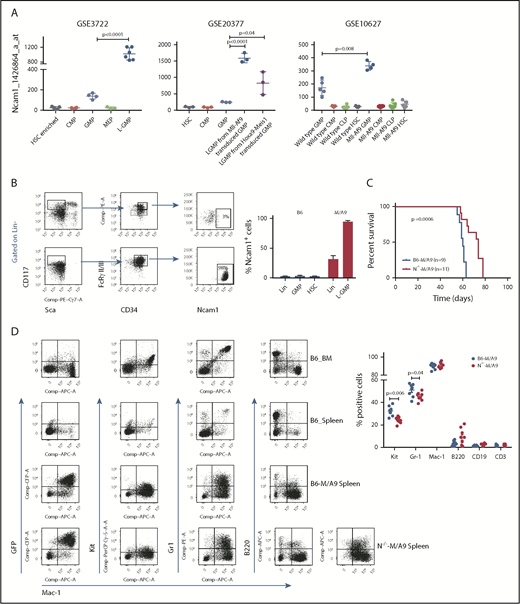
Ncam1 is highly expressed in murine leukemic Mll-Af9 stem cells. (A) Expression of Ncam1 was compared between normal hematopoietic and different leukemic progenitor populations in the indicated gene expression data sets. (B) Ncam1 cell surface expression in different normal and leukemic progenitor populations was analyzed by flow cytometry. Shown are scatter plots of representative samples (left) depicting the gating strategy to compare Ncam1 expression in GMP vs L-GMP. Right, Bar graphs showing the percentage of Ncam1 expression in indicated hematopoietic populations. Error bars represent mean of 3 biological replicates ± SD of mean. (C) Kaplan-Meier survival curves of recipient animals transplanted with equal amounts of B6-M/A9 or N−/−-M/A9 whole BM cells. Differences were compared by the log-rank test. (D) Representative scatter plots showing expression of GFP, c-Kit, Gr1, B220, and Mac1 in normal murine BM (B6_BM) and spleen (B6_Spleen) cells and B6-M/A9 and N−/−-M/A9 spleen cells (left). The percentage of positive labeled cells of B6-M/A9 and N−/−-M/A9 samples was compared by 2-tailed Student t test (right). Error bars represent mean of 8 biological replicates ± SD of mean.
Ncam1 is highly expressed in murine leukemic Mll-Af9 stem cells. (A) Expression of Ncam1 was compared between normal hematopoietic and different leukemic progenitor populations in the indicated gene expression data sets. (B) Ncam1 cell surface expression in different normal and leukemic progenitor populations was analyzed by flow cytometry. Shown are scatter plots of representative samples (left) depicting the gating strategy to compare Ncam1 expression in GMP vs L-GMP. Right, Bar graphs showing the percentage of Ncam1 expression in indicated hematopoietic populations. Error bars represent mean of 3 biological replicates ± SD of mean. (C) Kaplan-Meier survival curves of recipient animals transplanted with equal amounts of B6-M/A9 or N−/−-M/A9 whole BM cells. Differences were compared by the log-rank test. (D) Representative scatter plots showing expression of GFP, c-Kit, Gr1, B220, and Mac1 in normal murine BM (B6_BM) and spleen (B6_Spleen) cells and B6-M/A9 and N−/−-M/A9 spleen cells (left). The percentage of positive labeled cells of B6-M/A9 and N−/−-M/A9 samples was compared by 2-tailed Student t test (right). Error bars represent mean of 8 biological replicates ± SD of mean.
Ncam1 expression confers stress resistance and prevents LSC exhaustion
To gain insight into the functional role of aberrant Ncam1 expression in vivo, we transplanted lethally irradiated recipients with M/A9-transduced Lin− BM cells derived from Ncam1−/− or Ncam1wt/wt donor animals (hereafter N−/−-M/A9 and B6-M/A9, respectively). As shown in Figure 4C, depletion of Ncam1 significantly prolonged overall survival compared with B6-M/A9 recipient mice. Immunophenotype analysis demonstrated decreased expression of c-Kit and Gr-1 in N−/−-M/A9 cells (Figure 4D) hinting toward a less aggressive myeloid phenotype. In addition, we observed aberrant expression of the B-cell maker B220 in some samples, indicating biphenotypic leukemia and/or compensatory B220 expression in N−/−-M/A9 leukemia. In line with our cell line results, loss of Ncam1 significantly sensitized leukemic blasts to low-dose Ara-C treatment in vitro (supplemental Figure 5).
We next addressed the question of whether Ncam1 is involved in the regulation of LSC maintenance. In vitro, colony-forming cell (CFC) assays demonstrated reduced numbers of colonies in N−/−-M/A9 cells compared with B6-M/A9 controls, but replating activity was retained (Figure 5A). When replating intervals were extended to 10 days, going along with limited nutritional supply and increased cellular stress, self-renewal was gradually lost in N−/−-M/A9 cells (Figure 5B). Furthermore, we performed secondary transplantation into sublethally irradiated recipient mice. All transplanted mice eventually developed leukemia but disease latency was significantly prolonged in N−/−-M/A9 recipients (Figure 5C). Spleen size was substantially reduced compared with B6-M/A9 mice suggesting a less aggressive disease phenotype (Figure 5D). Again, we observed lower expression of the stem cell marker c-Kit in N−/−-M/A9 mice (Figure 5E) indicating a loss of LSCs.
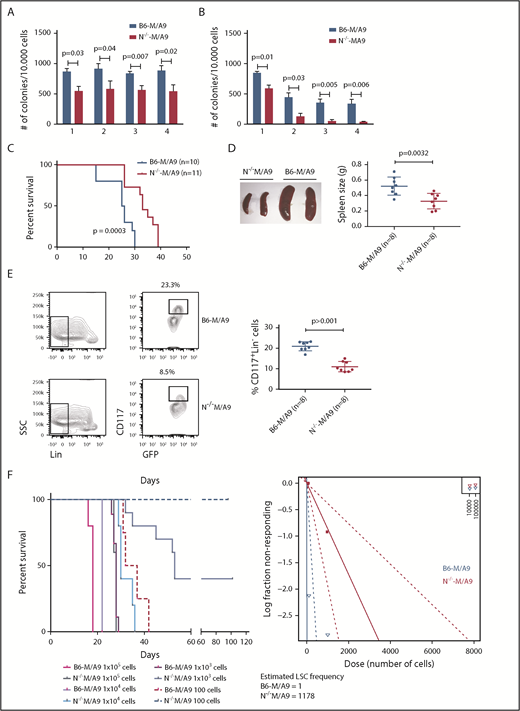
Ncam1 expression confers stress resistance and prevents LSC exhaustion. (A-B) Clonogenic assays using isogenic B6-M/A9 and N−/−-M/A9 cells. Replating was performed every 7 days (A) and every 10 days (B). The number of colonies was compared by 2-tailed Student t test. Error bars represent mean of 3 to 5 biological replicates ± SD of mean. (C) Kaplan-Meier survival curves of secondary transplanted animals (100 000 cells per mouse). Survival of B6-M/A9 (n = 10) or N−/−-M/A9 (n = 11) recipients was compared by the log-rank test. (D) Left, Representative examples of spleens derived from leukemic mice. Right, Statistics of spleen sizes derived from secondary transplanted B6-M/A9 or N−/−-M/A9 mice. (E) Left, representative scatter plots of CD117 expression; right, statistics showing the percentage of Lin−Kithigh B6-M/A9 (N = 8) or N−/−-M/A9 (N = 8) AML cells after secondary transplantation. (F) Limiting-dilution transplantation experiment. Animals were transplanted with different numbers of BM cells derived from secondary recipients as indicated (1 × 105: n = 3; all other groups: n = 5). Red/purple colors represent wild-type animals, blueish colors Ncam1-depleted animals. Left, Survival of each group is shown as Kaplan-Meier curve. The estimated LSC number was calculated by L-Calc (see “Materials and methods”). Left, Corresponding extreme limiting dilution analysis graph.
Ncam1 expression confers stress resistance and prevents LSC exhaustion. (A-B) Clonogenic assays using isogenic B6-M/A9 and N−/−-M/A9 cells. Replating was performed every 7 days (A) and every 10 days (B). The number of colonies was compared by 2-tailed Student t test. Error bars represent mean of 3 to 5 biological replicates ± SD of mean. (C) Kaplan-Meier survival curves of secondary transplanted animals (100 000 cells per mouse). Survival of B6-M/A9 (n = 10) or N−/−-M/A9 (n = 11) recipients was compared by the log-rank test. (D) Left, Representative examples of spleens derived from leukemic mice. Right, Statistics of spleen sizes derived from secondary transplanted B6-M/A9 or N−/−-M/A9 mice. (E) Left, representative scatter plots of CD117 expression; right, statistics showing the percentage of Lin−Kithigh B6-M/A9 (N = 8) or N−/−-M/A9 (N = 8) AML cells after secondary transplantation. (F) Limiting-dilution transplantation experiment. Animals were transplanted with different numbers of BM cells derived from secondary recipients as indicated (1 × 105: n = 3; all other groups: n = 5). Red/purple colors represent wild-type animals, blueish colors Ncam1-depleted animals. Left, Survival of each group is shown as Kaplan-Meier curve. The estimated LSC number was calculated by L-Calc (see “Materials and methods”). Left, Corresponding extreme limiting dilution analysis graph.
To further test this hypothesis, we performed tertiary, limited dilution transplantation assays. All B6-M/A9 recipient mice succumbed to leukemia independent of transplanted cell numbers (Figure 5F). In contrast, whereas all N−/−-M/A9 recipient mice transplanted with 1 × 105 or 1 × 104 cells developed AML with increased disease latency, 37% and 100% of mice transplanted with 1 × 103 or 1 × 102 cells, respectively, survived. Calculation of stem cell numbers demonstrated a significant reduction of LSC frequency in Ncam1-depleted leukemia compared with controls (1 of 1178 vs 1 of 1, respectively). In conclusion, our data provide evidence that Ncam1 is highly expressed in LSCs and is involved in the maintenance of LSCs.
MEK-ERK and glycolysis are crucial signaling pathways in NCAM1+ AML
To identify potential mechanisms involved in the observed functional properties related to NCAM1 expression, we performed stable isotope labeling with phosphoproteomic analyses in NOMO-1 cells either expressing shNCAM_2 or scrambled shRNA followed by mass spectrometry. A total of 1893 phosphosites representing 663 proteins were differentially regulated upon NCAM1 knockdown (Figure 6A). Suppression of NCAM1 was followed by decreased phosphorylation of the SRC kinases FYN and LYN, RICTOR, RAPTOR, and several central members of the RAS-RAF-MAPK signaling pathway, such as extracellular signal-regulated kinase 1/2 (ERK1/2), MEK1/2, CRKL, FOSL2, JUNB, CREB1, and MYC (Figure 6A). Immunoblot analysis confirmed suppressed phosphorylation of MEK1/2 and ERK1/2, and also of the p38 kinase after NCAM1 knockdown in NOMO1 and SKM1 cells (supplemental Figure 6A). Of note, knockdown of NCAM1 suppressed MEK-ERK signaling despite the fact that NOMO-1 cells harbor mutated KRAS linked to constitutive activation of this pathway indicating the requirement of upstream regulators as described for mutant KRAS.43,44 To overcome MAPK-ERK activation mediated by FLT3-ITD in MOLM-14 cells, combined treatment with the FLT3 inhibitor crenolanib and NCAM1 knockdown was required (supplemental Figure 6A). Overrepresentation analysis of gene ontology (GO) terms identified biological processes related to cell cycle regulation, apoptosis, and cell morphology (Figure 6B). Furthermore, we observed differential regulation of tyrosine phosphorylation in biological processes involved in the regulation of glucose transport (Figure 6B), which has previously been linked to the MEK-ERK signaling.45-47 Aiming to further characterize the effects of the NCAM1 perturbation in highly heterogeneous AML cell lines, we next performed RNA-seq in NOMO-1, MOLM-14, and SKM-1 cells stably expressing either shNCAM_2 or scrambled shRNA. We first analyzed REACTOME48 and KEGG49 term overrepresentation of significantly decreased transcripts in shNCAM_2 cells. Glucose metabolism/glycolysis was most significantly overrepresented among all terms in both REACTOME and KEGG (supplemental Figure 6B). Finally, analyzing gene set enrichment with gene set enrichment analysis (GSEA)50 for all expressed transcripts, we confirmed decreased expression of genes involved in glucose metabolism after NCAM1 knockdown (Figure 6C). Of note, attenuated glycolysis was accompanied by significantly reduced expression of hypoxia target genes (Figure 6C).
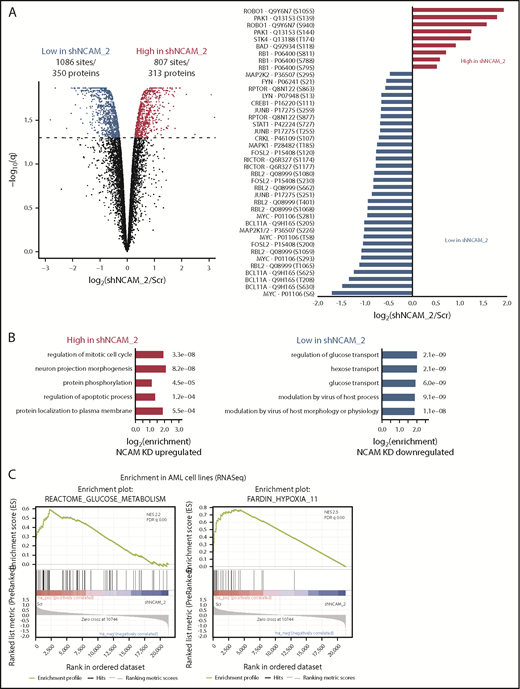
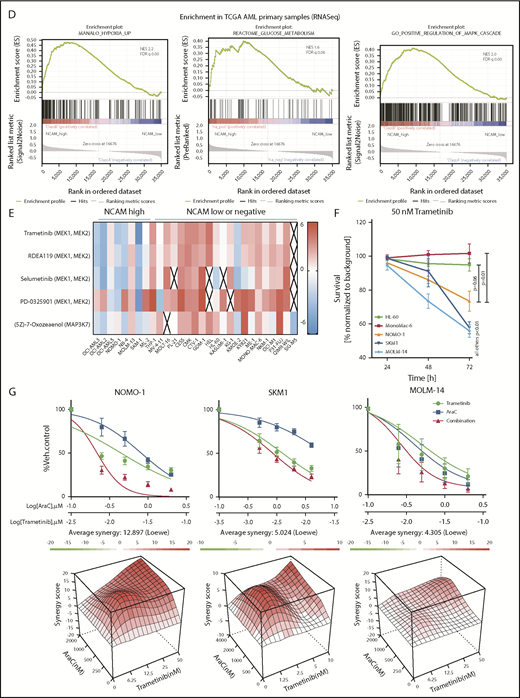
MEK-ERK signaling is a crucial pathway and a therapeutic target in NCAM+ AML. (A) Quantitative phosphoproteomics of stable isotope labeled Scr- and shNCAM_2-expressing NOMO-1 cells. Left, Volcano plot displaying differential phosphorylation sites of the phosphoproteomic data. Right, Selected differential phosphorylation sites that have a documented impact on the protein function. Data were provided from 3 independent experiments. Statistical analysis was performed with the Limma package in R. (B) List of the 5 most enriched GO biological processes (GO BP) terms for enriched phosphorylation in the indicated groups. (C) RNA-seq was performed after Dox-mediated induction of scrambled shRNA or shNCAM_2 in MOLM-14, SKM1 and NOMO-1 cells for 72 hours and gene set enrichment analysis was performed using the GSEA preranked on all the expressed transcripts. Shown are the gene signatures that most significantly decrease after NCAM downregulation. (D) Samples of the TCGA dataset were preselected for the top (NCAM1_high) and bottom (NCAM1_low) quintile of NCAM1 expression. Gene set enrichment analysis was performed using GSEA preranked on all expressed genes in NCAM1_high and -low samples and compared for overlapping enrichment with the curated MSigDB data sets. Shown are selected enrichments based on the terms shown in panels A-C. (E) Heatmap of sensitivity of leukemic cell lines upon treatment with different small-molecule inhibitors that target the MEK-ERK signaling pathway. Analysis was performed on data derived from the Genomic in cancer sensitivity51 project. (F) AML cell lines were treated with Trametinib (50 nM) for 24, 48, and 72 hours as indicated. Cellular proliferation was assessed by MTT absorbance and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (G) NOMO-1, SKM-1 and MOLM-14 cells were treated with Ara-C, Trametinib or in combination for 48 hours. Cell viability was measured by MTT, while treatment synergy was measured using the Loewe model (see “Materials and methods”). Shown are dose-response curves in the top panels and 3-dimensional diffusion plots demonstrating treatment synergism in the bottom panels.
MEK-ERK signaling is a crucial pathway and a therapeutic target in NCAM+ AML. (A) Quantitative phosphoproteomics of stable isotope labeled Scr- and shNCAM_2-expressing NOMO-1 cells. Left, Volcano plot displaying differential phosphorylation sites of the phosphoproteomic data. Right, Selected differential phosphorylation sites that have a documented impact on the protein function. Data were provided from 3 independent experiments. Statistical analysis was performed with the Limma package in R. (B) List of the 5 most enriched GO biological processes (GO BP) terms for enriched phosphorylation in the indicated groups. (C) RNA-seq was performed after Dox-mediated induction of scrambled shRNA or shNCAM_2 in MOLM-14, SKM1 and NOMO-1 cells for 72 hours and gene set enrichment analysis was performed using the GSEA preranked on all the expressed transcripts. Shown are the gene signatures that most significantly decrease after NCAM downregulation. (D) Samples of the TCGA dataset were preselected for the top (NCAM1_high) and bottom (NCAM1_low) quintile of NCAM1 expression. Gene set enrichment analysis was performed using GSEA preranked on all expressed genes in NCAM1_high and -low samples and compared for overlapping enrichment with the curated MSigDB data sets. Shown are selected enrichments based on the terms shown in panels A-C. (E) Heatmap of sensitivity of leukemic cell lines upon treatment with different small-molecule inhibitors that target the MEK-ERK signaling pathway. Analysis was performed on data derived from the Genomic in cancer sensitivity51 project. (F) AML cell lines were treated with Trametinib (50 nM) for 24, 48, and 72 hours as indicated. Cellular proliferation was assessed by MTT absorbance and compared by 2-tailed Student t test. Error bars represent mean of 3 measurements ± SD of mean. (G) NOMO-1, SKM-1 and MOLM-14 cells were treated with Ara-C, Trametinib or in combination for 48 hours. Cell viability was measured by MTT, while treatment synergy was measured using the Loewe model (see “Materials and methods”). Shown are dose-response curves in the top panels and 3-dimensional diffusion plots demonstrating treatment synergism in the bottom panels.
To confirm the relevance of these findings in an AML patient cohort, we compared transcriptional profiles of patient samples with high (top quintile) and low (bottom quintile) NCAM1 expression levels derived from the TCGA AML RNA-seq data set.36 In line with our knockdown experiments, GSEA analysis demonstrated enrichment for the MAPK cascade, glucose metabolism, and hypoxia gene targets in NCAM1 high samples (Figure 6D).
MEK-ERK signaling is a therapeutic target in NCAM1+ AML
To assess potential effects of MEK-ERK inhibition, we reviewed a large-scale cancer cell line compound database and linked drug response with NCAM1 expression.51 As shown in Figure 6E, cell lines with high NCAM1 expression levels are specifically sensitive to MEK1/2- and MAPK3K7-inhibitors, whereas most NCAM1− cell lines were resistant. To confirm these data, we treated several AML cell lines with trametinib, a specific MEK1/2 inhibitor. MEK1/2 inhibition specifically suppressed cell growth of NCAM1-expressing cell lines, whereas NCAM1− cells did not respond (Figure 6F). As our previous data demonstrated that NCAM1 suppression sensitized AML cells to chemotherapy, we investigated the effect of Ara-C in combination with trametinib. Combined treatment with Ara-C and trametinib caused synergistic inhibition of cell growth and significantly increased apoptotic cell death compared with single-agent therapies in NOMO-1, SKM-1, and MOLM-14 cell lines (Figure 6G; supplemental Figure 6C). In contrast, no additional effects were observed in NCAM1− cells (supplemental Figure 6C). Altogether, these data demonstrate that NCAM1 acts, at least partially, through the MAPK signaling pathway and that MEK1/2 inhibition provides an interesting therapeutic option to overcome NCAM1-mediated resistance against chemotherapy.
Discussion
In this study, we investigated the biological function of aberrant NCAM1 expression in AML. Using different but complementary genetic strategies, our data demonstrate an essential role of NCAM1 as a regulator of survival, stress resistance, and self-renewal of AML blasts in vitro and in vivo.
As previously shown, NCAM1 was highly expressed in t(8;21)-mutant AML.24 In addition, NCAM1 expression was spread across a variety of cytogenetically and molecularly defined AML categories. Within all these subsets, NCAM1 expression was highly heterogeneous, indicating an unspecific mechanism of regulation of gene expression. Indeed, several transcription factors such as MEIS1, MEF2, members of the STAT family, and the p48-isoform of RUNX130 are involved in the regulation of NCAM1 expression, which suggests the presence of a rewired epigenetic landscape providing access of already available transcription factors to the NCAM1 promoter. Alternatively, the heterogeneous expression of NCAM1 might be related to the cell of origin during malignant transformation. In murine Mll-Af9 gene expression data, high Ncam1 expression was demonstrated in LSCs derived from normal GMPs but not upon transduction of HSCs.52 In our experiments, whole BM was used as a source for viral transduction, suggesting that the major target population consisted of GMPs. Of note, increased Ncam1 expression associates with the murine GMP signature,42 which supports our hypothesis that the cell of origin of leukemic transformation represents a prerequisite for aberrant NCAM1 expression.
NCAM1 (CD56) is widely used as marker to monitor minimal residual disease,53-55 however, the biological function of this cell surface protein has not been addressed yet. Our data provide evidence that AML cells become addicted to NCAM1 expression. Serial transplantation of M/A9-transformed Ncam1−/−-BM cells leads to a gradual loss of leukemia initiating cells compared with wild-type cells. Moreover, knockdown of NCAM1 sensitizes AML blasts to genotoxic agents and targeted therapies, whereas overexpression in NCAM1− cell lines enhances resistance to therapy. In line with these results, aberrant NCAM1 expression is associated with worse overall survival and high relapse rates.24-26 To further explore the mechanism behind the observed phenotype, we performed global phosphoproteome and transcriptome analyses. NCAM1 expression was associated with key signatures affecting the regulation of carbohydrate metabolism and hypoxia. Furthermore, the presence of NCAM1 caused strong activation of the MEK-ERK signaling pathway and posttranslational modification of effector proteins such as MYC, c-JUN, and CREB1.56 NCAM1-mediated activation of the MEK-ERK pathway has also been described in neural development.19,57,58 In these studies, MEK-ERK signaling was dependent on the homophilic binding of different NCAM1 molecules or the heterophilic interaction with FGFR followed by FYN-FAK activation.19,57,58 In line with these data, we observed decreased phosphorylation of FYN upon NCAM1 knockdown; however, steady state expression levels of PTK2 (FAK) were rarely detectable in several NCAM1+ AML cell lines (data not shown). Moreover, pharmaceutical inhibition of FGFR in various NCAMhigh AML cell lines had no effect on cell survival, either alone or in combination with chemotherapy (data not shown). Although FGFR has been described as a major binding partner, NCAM1 can interact with several other cell surface complexes and activates a variety of different downstream molecules.16 Alternatively, NCAM1 can act as a reservoir for cytokines, chemokines, and growth factors. Due to its zipper-like structure, established by homophilic cis and trans binding and further amplified by posttranslational polysialylation,12,59 NCAM1 traps surrounding cytokines and could provide continuous supply to nearby cell surface receptors.
Although the exact mechanism of NCAM1-mediated downstream signaling remains unclear, the strong activation of the MEK-ERK pathway might represent an interesting therapeutic strategy in NCAM1+ leukemia. Indeed, treatment with the MEK-inhibitor trametinib caused inhibition of cell growth and apoptotic cell death in combination with chemotherapy. Treatment response was only observed in NCAM1+ AML cell lines and was independent of the presence of NRAS or KRAS mutations (see supplemental Table 1). Although in early clinical trials effects of MEK inhibitors60 were particularly observed in RAS-mutant leukemias, the expression of NCAM1 might serve as an alternative biomarker and requires further investigation.
Global sequencing efforts in primary AML patient samples identified that ∼60% harbor mutations in genes of oncogenic signaling pathways.36 The absence of oncogenic mutations is often compensated by overexpression of receptor tyrosine kinases with well-established functions in normal hematopoiesis such as cKIT, interleukin-3 receptor, or FLT3.61-63 Our data provide evidence that aberrant expression of otherwise not-myelopoiesis-related cell surface proteins can induce mitogenic signaling pathways and also regulate cellular homeostasis in leukemic cells.
The RNASeq data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE123071).
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kerstin Kunz and Birgit Enders for technical assistance.
This work was supported by the German Research Foundation (KI1100-4-1) and intramural funding of the University Medical Center of Mainz. D.S. was a Postdoctoral Fellow of the Dr Mildred Scheel Foundation (111875). L.B. was supported in part by the German Research Foundation (Heisenberg-Stipendium BU 1339/3-1). P.B. was supported by the Emmy Noether Program (BE 5342/1-1), SFB1177 on selective autophagy from the German Research Foundation, and a Marie Curie Integration grant from the European Commission (630763).
Authorship
Contribution: D.S. designed experiments, performed research, analyzed data, and wrote the manuscript; A.S. and V.S. designed experiments, performed research, and analyzed data; J.S., J.H., P.S.H., A.D., and W.H.G. performed research and analyzed data; O.K. and E.-M.F. analyzed data; G.R., C.S., M.T., and L.B. designed experiments; P.B. designed experiments and analyzed data; and T.K. designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thomas Kindler, Department of Hematology, Medical Oncology and Pneumology, University Medical Center, Mainz, Germany; e-mail: thomas.kindler@unimedizin-mainz.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal