

Key Points
CD1a CARTs exhibit specific and robust cytotoxicity in vitro and in vivo using both T-ALL cell lines and primary coT-ALL cells.
CD1a CARTs are fratricide resistant and exhibit long-term persistence in vivo with antileukemic activity in rechallenge experiments.

Abstract
Relapsed/refractory T-cell acute lymphoblastic leukemia (T-ALL) has a dismal outcome, and no effective targeted immunotherapies for T-ALL exist. The extension of chimeric antigen receptor (CAR) T cells (CARTs) to T-ALL remains challenging because the shared expression of target antigens between CARTs and T-ALL blasts leads to CART fratricide. CD1a is exclusively expressed in cortical T-ALL (coT-ALL), a major subset of T-ALL, and retained at relapse. This article reports that the expression of CD1a is mainly restricted to developing cortical thymocytes, and neither CD34+ progenitors nor T cells express CD1a during ontogeny, confining the risk of on-target/off-tumor toxicity. We thus developed and preclinically validated a CD1a-specific CAR with robust and specific cytotoxicity in vitro and antileukemic activity in vivo in xenograft models of coT-ALL, using both cell lines and coT-ALL patient–derived primary blasts. CD1a-CARTs are fratricide resistant, persist long term in vivo (retaining antileukemic activity in re-challenge experiments), and respond to viral antigens. Our data support the therapeutic and safe use of fratricide-resistant CD1a-CARTs for relapsed/refractory coT-ALL.
Introduction
T-cell lineage acute lymphoblastic leukemia (T-ALL) is a malignant disorder resulting from leukemic transformation of thymic T-cell precursors.1 T-ALL is phenotypically and genetically heterogeneous and is commonly associated with genetic alterations/mutations in transcription factors involved in hematopoietic stem and progenitor cell (HSPC) homeostasis and in master regulators of T-cell development.2 T-ALL comprises 10% to 15% and 20% to 25% of all acute leukemias diagnosed in children and adults, respectively,3,4 with a median diagnostic age of 9 years.5-7 Intensive chemotherapy regimens have led to the improved survival of patients with T-ALL; however, the event-free and overall (OS) survival remains <70%, and relapsed/refractory (R/R) T-ALL has a particularly poor outcome. There are currently no potential curative options beyond hematopoietic cell transplantation and conventional chemotherapy, which is linked to large trade-offs in toxicities,4,8 reinforcing the need for novel targeted therapies.
Immunotherapy has generated unprecedented expectations in cancer treatment and relies on the immune system as a powerful weapon against cancer. In recent years, adoptive cellular immunotherapy based on chimeric antigen receptors (CARs) has shown great potential. CAR therapy redirects genetically modified T cells to specifically recognize and eliminate specific antigen-expressing tumor cells in a major histocompatibility complex–independent fashion.9,10 The success of CAR T cells (CARTs) redirected against CD19 or CD22 is now indisputable for B-cell malignancies (mainly B-ALL).11-14 However, strategies targeting T-cell malignancies using CARTs remain challenging because of the shared expression of target antigens between CARTs and T-lineage tumoral cells. In this regard, CARTs against pan T-cell antigens have 2 major drawbacks: (1) CARTs self-targeting/fratricide; and (2) T-cell aplasia, leading to life-threating immunodeficiency.15-17
Recent elegant preclinical studies showed that T cells transduced with either CD7, CD3, CD5, or T-cell receptor CARs (the most expressed pan T-cell antigens) efficiently eliminate T-ALL blasts in vitro and are able to control the disease in vivo,15-20 leading very recently to pioneering phase 1 clinical trials with CARTs for T-ALL (#NCT03690011 and #NCT03590574). However, innovative approaches, such as CRISPR/Cas9 genome editing or protein expression blockers, seem needed for disruption of the target antigen in T cells before CAR transduction to avoid extensive self-antigen–driven fratricide.15-17,19
Gene expression profiling and multicolor immunophenotyping classify T-ALLs into distinct subgroups that mostly reflect a particular stage of differentiation arrest.21 Cortical T-ALL (coT-ALL) is a major subgroup of T-ALL characterized by the surface expression of CD1a, consistent with a developmental arrest at the cortical stage.22-24 There are 4 CD1 isoforms (CD1a, CD1b, CD1c, and CD1d) in humans, whereas only the CD1d isoform is expressed in the mouse.25 Upon recognition of the CD1 ligand complex by the T-cell receptor, CD1-dependent T cells are activated in a variety of immunological contexts. Loss-of-function studies revealed that CD1-deficient mice may be more susceptible to some viruses, bacteria, and protozoa.26,27 Unfortunately, the role of CD1 isoforms in human infection remains elusive. CD1a is a lipid-presenting molecule whose expression is essentially restricted to coT-ALL and Langerhans cell (LC) histiocytosis and is practically absent in human tissues with the exception of developing cortical thymocytes and LC.28,29 Here, we tested the feasibility of targeting CD1a+ coT-ALL using CD1a CARTs. We report that CD1a-specific CARTs exhibit robust cytotoxicity against CD1a+ coT-ALL cell lines and primary coT-ALL cells, both in vitro and in vivo. CD1a CARTs are fratricide resistant and remain functional in vivo after 13 weeks, as shown by leukemia rechallenge experiments. Fratricide-resistant CD1a CARTs thus represent a safe and innovative adoptive immunotherapy for coT-ALL and potentially for other CD1a+ tumors such as LC histiocytosis.
Methods
CD1a-scFv generation and CAR design
The CD1a-specific single-chain variable fragment (scFv) derived from the NA1/34.HLK clone of CD1a-specific antibody was obtained by using commercial synthesis (MilliporeSigma) with the mouse IgG Library Primer Set (Progen). It was cloned into a pCCL lentiviral-based second-generation CAR backbone containing a human CD8 transmembrane domain, human 4-1BB and CD3z endodomains, and a T2A–green fluorescent protein (GFP) cassette. Identical lentiviral vectors expressing either GFP alone (MOCK) or CD22 CAR backbone were used as controls.
CAR-expressing lentiviral production, T-cell transduction, activation, and expansion
CAR-expressing viral particles pseudotyped with vesicular stomatitis virus G glycoprotein were generated in 293T cells by using standard polyethylenimine transfection protocols and concentrated by ultracentrifugation, as previously described.30 Viral titers were consistently in the range of 108 TU/mL. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats from healthy volunteers by using Ficoll-Hypaque gradient centrifugation. Buffy coats were obtained from the Barcelona Blood and Tissue Bank upon institutional review board approval (HCB/2018/0030). T cells were activated by plate-bound anti-CD3 (OKT3) and anti-CD28 (BD Biosciences) antibodies for 2 days and were then transduced with CAR-expressing lentivirus (multiplicity of infection = 10) in the presence of interleukin-7 (IL-7) and IL-15 (10 ng/mL; Miltenyi Biotec).16,18 The cell surface expression of CD1aCAR was traced by fluorescence-activated cell sorting (FACS) expression of GFP and by using AffiniPure F(ab')2 Fragment Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch Laboratories). Proper activation of CAR-transduced T cells was shown by staining for CD25 and CD69 after 2-day expansion.
Immunophenotyping of healthy CD34+ progenitors, T cells, and primary T-ALL samples
The expression of CD1a antigen in CD34+ HSPCs, CD34+CD7+ thymic T-cell progenitors, and CD3+ T cells was prospectively analyzed in fresh human thymus, fetal liver and bone marrow (BM), cord blood, and adult BM and peripheral blood (PB) (n = 3). Fetal tissue was collected as previously described31,32 from developing embryos aborted at 18 to 22 weeks of pregnancy, obtained from the MRC/Wellcome Trust Human Developmental Biology Resource upon informed consent and approval by our local ethics committee (CMRB-CEIC-26/2013). Neonatal and adult tissues were obtained from the Barcelona Blood and Tissue Bank upon institutional review board approval (HCB/2018/0030). Primary T-ALL samples and diagnostic immunophenotyping data (n = 38) were obtained from the Spanish hospitals Sant Joan de Déu, Germans Trias i Pujol, Santa Creu i San Pau (Barcelona), Niño Jesús (Madrid), and Virgen de la Arrixaca (Murcia). Supplemental Table 1 (available on the Blood Web site) shows the main clinical-biological features of the CD1a++ coT-ALL cases. For immunophenotyping of T-ALL primary samples, the following fluorochrome-conjugated monoclonal antibodies (mAbs) were used: anti-CD2-PE, CD7-FITC/PE, CD13-PerCP-Cy5.5, CD34-APC, CD3-PE, CD5-FITC, CD4-BV-421, CD8-APC-Cy7, CD45-AmCyan, CD1a-BV-421/APC/PE, CD33-APC, and CD123-APC (BD Biosciences). Isotype-matched, nonreactive fluorochrome-conjugated mAbs were always used as a fluorescence reference. Briefly, 5 × 105 PBMCs were incubated with erythrocyte-lysing solution (BD Biosciences) for 10 minutes and then stained with mAbs (20 minutes at 4°C in the dark). Stained cells were washed in phosphate-buffered saline and FACS-analyzed on a FACSCanto-II cytometer equipped with FACSDiva software (BD Biosciences).33-35 CD1a antigen density was determined by using BD QuantiBRITE PE, as described elsewhere.36
In vitro cytotoxicity assays and cytokine release determination
The Jurkat, MOLT4, and NALM6 cell lines were purchased from DSMZ and expanded according to DSMZ recommendations. Luciferase (Luc)/GFP-expressing cells were stably generated by retroviral transduction and FACS purification of GFP-positive cells.37 Target cells (cell lines and primary T-ALL blasts) were labeled with 3 µM eFluor 670 (eBioscience) and incubated with CD1a, CD22, or MOCK CARTs at different effector:target (E:T) ratios for the indicated time periods. CART-mediated cytotoxicity was determined by analyzing the residual alive (7-amino actinomycin D negative) eFluor 670–positive target cells at each time point and E:T ratio. Absolute cell counts were determined by using Trucount absolute count beads (BD Biosciences). In addition, FACS-sorted CD3+CD1a– mature T cells from PB of patients with coT-ALL at presentation were activated, transduced with CD1a CAR, and tested against their eFluor 670–labeled autologous-matched CD1a+ coT-ALL blasts. Leukemic blasts were never FACS-sorted in primary samples. The production of the proinflammatory cytokines IL-2, tumor necrosis factor α (TNFα), and interferon γ (IFN-γ) was measured by using an enzyme-linked immunosorbent assay (BD Biosciences) in supernatants harvested after 16 hours.
In vivo Jurkat and T-ALL patient-derived xenograft models
Six- to 12-week-old nonobese diabetic-Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) were bred and housed under pathogen-free conditions in the animal facility of the Barcelona Biomedical Research Park. Mice were irradiated (2 Gy) and IV transplanted with 3 × 106 Luc/GFP–expressing Jurkat cells or with 1 × 106 primary cortical CD1a+ T-ALL blasts (primary and primograft-expanded).38 Between 1.5 and 5 × 106 CD1a or MOCK CARTs were IV infused 3 days later. When Luc/Jurkat cells were used, tumor burden was followed by bioluminescence (BLI) using the Xenogen IVIS 50 Imaging System (PerkinElmer). To measure luminescence, mice were given 150 mg/kg of D-luciferin intraperitoneally, and tumor burden was monitored at the indicated time points. Living Image software (PerkinElmer) was used to visualize and calculate total luminescence. Tumor burden of primary coT-ALL samples was followed up by biweekly bleeding and FACS analysis. Mice were euthanized when MOCK CART-treated animals were leukemic, and tumor burden (hHLA-ABC+hCD45+hCD3–/lowhCD1a+ graft) and effector T persistence (hHLA-ABC+hCD45+hCD3+hCD1a–GFP+) was analyzed in BM, PB, and spleen according to FACS. In rechallenge experiments, leukemia-free animals that had received an infusion of CD1a CARTs 5 to 7 weeks before were re-infused with either 1.5 × 106 Luc/Jurkat cells or 1 × 106 CD1a+ coT-ALL primary cells, and disease reappearance was followed up by BLI and FACS. All procedures were performed in compliance with the institutional animal care committee of the Barcelona Biomedical Research Park (DAAM7393).
ELISpot
Enzyme-linked immunospot assay (ELISpot) plates (MilliporeSigma) were coated with anti-human IFN-γ antibody (1-D1K; Mabtech) and kept overnight at 4°C. Plates were then washed 6 times with PBS containing 1% fetal calf serum, and cells from 3 independent donors were then plated at 5 to 10 × 105 cells/well and cultured in triplicate for 20 hours at 37°C and 5% carbon dioxide. We measured IFN-γ–secreting cells in response to CEF at 1 μg/mL, a peptide pool of T-cell epitopes of cytomegalovirus (CMV), Epstein-Barr virus (EBV), and flu virus, and to staphylococcal enterotoxin B at 1 μg/mL as a positive control. Plates were then revealed with biotinylated anti-human IFN-γ and streptavidin-alkaline phosphatase (Mabtech), as previously described.39,40 The frequency of IFN-γ–secreting cells was quantified by using ImmunoCapture and ImmunoSpot software (both, Cellular Technology Limited) to calculate the number of IFN-γ spot-forming units per 105.
Statistical analysis
Data from at least 3 individual donors are shown in all figures, and experimental duplicates were always performed. At least 5 animals were used in each in vivo condition. All P values were calculated by using the unpaired 2-tailed Student t test with Prism software (GraphPad Software). OS of mice was determined by using a Mantle-Cox test. P < .05 was considered statistically significant.
Results
CD1a specifically marks coT-ALL blasts
The shared expression of target antigens between CARTs and T-lineage blasts has limited immunotherapy approaches in T-ALL due to CART fratricide and potential life-threating T-cell aplasia. CD1a antigen was expressed in coT-ALLs, a major subset of T-ALLs (Figure 1A-B; supplemental Figure 1), and retained at relapse (Figure 1C). Similar to previous studies,41-43 in our cohort of patients with T-ALL (n = 38), 75% (n = 29) were phenotypically defined as CD1a+ coT-ALL; however, the expression of CD1a was homogeneous in only 50% (19 of 38) of the patients with T-ALL. Importantly, CD1a was absent in T cells in all extrathymic tissues (Figure 1D),28 and steady-state CD34+ HSPCs also lacked CD1a expression in multiple hematopoietic sites across ontogeny. T-cell development was initiated within the thymus by a first colonizing CD34highCD7–CD1a– primitive HSPC with lymphomyeloid potential, which then differentiated in response to the thymic microenvironment into CD34highCD7+CD1a– early T-cell progenitors (ETP).44 As thymic differentiation progressed, ETPs maintained CD7 expression and gradually lost CD34 expression, whereas CD1a expression emerged and was transiently confined to cortical thymocytes45 (Figure 1E). Within the CD34+ thymic HSPC population, ∼50% is represented by precortical CD34highCD7+CD1a– ETPs (Figure 1F, blue cells), allowing us to hypothesize that CD1a may be a feasible and safe immunotherapeutic target for R/R coT-ALL.3,41,46,47
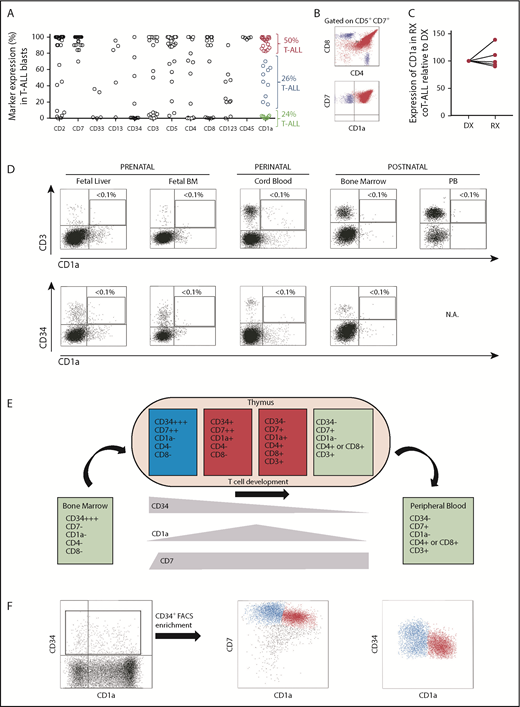
CD1a expression in T-ALL and normal hematopoiesis and thymopoiesis. (A) Immunophenotype of de novo T-ALL samples (n = 38) for the indicated markers. Red and blue circles identify CD1a+/++ and CD1alow/+ coT-ALL patients, respectively. Green circles depict non-coTALL patients. (B) Representative FACS dot plot of a patient with coT-ALL. Red cells are CD7+CD1a+ coT-ALL blasts, and blue cells are normal mature T cells (CD3+CD7+CD1a–, either CD4+ or CD8+) present in the diagnostic (DX) sample. (C) CD1a is retained at relapse (RX) (n = 5 DX-RX coT-ALL pairs). Data shown as CD1a expression in RX samples relative to the DX-matched samples (DX shown as 100% expression). (D) T cells and CD34+ HSPCs do not express CD1a across ontogeny. (E) Scheme depicting the phenotype of developing thymic T-cell populations. (F) Representative FACS for precortical (CD34highCD7++CD1a–) and cortical (CD34+CD7++CD1a+) thymocytes.
CD1a expression in T-ALL and normal hematopoiesis and thymopoiesis. (A) Immunophenotype of de novo T-ALL samples (n = 38) for the indicated markers. Red and blue circles identify CD1a+/++ and CD1alow/+ coT-ALL patients, respectively. Green circles depict non-coTALL patients. (B) Representative FACS dot plot of a patient with coT-ALL. Red cells are CD7+CD1a+ coT-ALL blasts, and blue cells are normal mature T cells (CD3+CD7+CD1a–, either CD4+ or CD8+) present in the diagnostic (DX) sample. (C) CD1a is retained at relapse (RX) (n = 5 DX-RX coT-ALL pairs). Data shown as CD1a expression in RX samples relative to the DX-matched samples (DX shown as 100% expression). (D) T cells and CD34+ HSPCs do not express CD1a across ontogeny. (E) Scheme depicting the phenotype of developing thymic T-cell populations. (F) Representative FACS for precortical (CD34highCD7++CD1a–) and cortical (CD34+CD7++CD1a+) thymocytes.
CD1a-redirected T cells (CD1a CARTs) expand without T-cell fratricide
We designed a second-generation CD1a CAR consisting of anti-CD1a scFv, a CD8 transmembrane spacer, and intracellular signaling domains from 4-1BB and CD3z coupled in-frame with GFP through a T2A sequence (Figure 2A). The expression of the CD1a CAR was easily detected through coexpression of both scFv and GFP in 293T cells (Figure 2B) and in primary CD4+ and CD8+ T-cell subsets (Figure 2C). Importantly, activated (CD69+CD25+) CD1a CARTs (Figure 2D) continuously expanded 200-fold over a 12-day period, similar to MOCK T cells (Figure 2E), showing that redirecting CARTs against CD1a antigen does not induce T-cell fratricide.
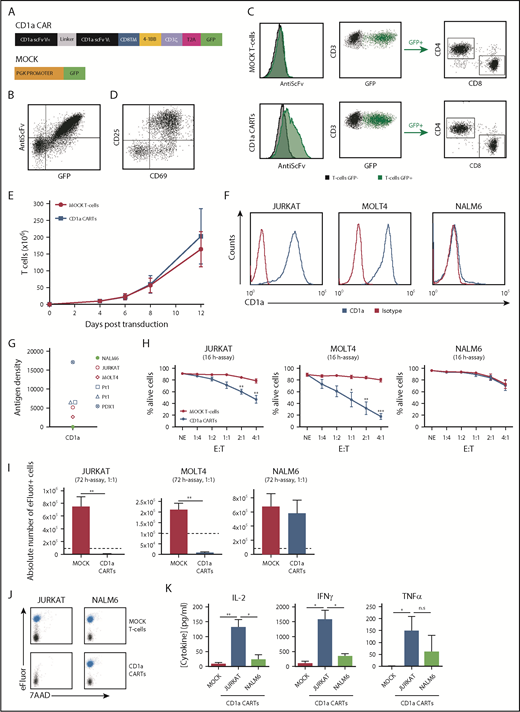
CD1a CARTs specifically target and eliminate CD1a+ T-ALL cell lines in vitro. (A) Scheme of the CD1aCAR construct used. (B) CAR detection in 293T cells using an anti-scFv mAb and GFP. (C) Representative CAR transduction and detection in CD4+ and CD8+ T cells (n = 6). (D) Proper T-cell activation (n = 3). (E) Robust expansion of activated T cells transduced with either MOCK or CD1a CAR reveals no signs of fratricide (n = 4). (F) Surface expression of CD1a (blue line) in Jurkat, MOLT4, and NALM6 cell lines. (G) CD1a antigen density in cell lines, primary coT-ALL samples, and primografts. (H) Cytotoxicity of CD1a CARTs and MOCK T cells against coT-ALL and B-ALL cell lines at the indicated E:T ratios in 16-hour assays (n = 4). (I) Absolute counts of alive eFluor-positive target cells measured according to FACS in 72-hour cytotoxicity assays at a 1:1 E:T ratio. (J) Representative FACS analysis of cytotoxicity with target cells labeled with eFluor 670 (shown in blue). (K) Enzyme-linked immunosorbent assay showing high-level production of the inflammatory cytokines IL-2, IFN-γ, and TNFα by CD1a CARTs exposed to Jurkat and NALM6 (negative control) cells in 16-hour assays at a 1:1 E:T ratio (n = 4). n.s., not significant. *P < .05, **P < .01, ***P < .001.
CD1a CARTs specifically target and eliminate CD1a+ T-ALL cell lines in vitro. (A) Scheme of the CD1aCAR construct used. (B) CAR detection in 293T cells using an anti-scFv mAb and GFP. (C) Representative CAR transduction and detection in CD4+ and CD8+ T cells (n = 6). (D) Proper T-cell activation (n = 3). (E) Robust expansion of activated T cells transduced with either MOCK or CD1a CAR reveals no signs of fratricide (n = 4). (F) Surface expression of CD1a (blue line) in Jurkat, MOLT4, and NALM6 cell lines. (G) CD1a antigen density in cell lines, primary coT-ALL samples, and primografts. (H) Cytotoxicity of CD1a CARTs and MOCK T cells against coT-ALL and B-ALL cell lines at the indicated E:T ratios in 16-hour assays (n = 4). (I) Absolute counts of alive eFluor-positive target cells measured according to FACS in 72-hour cytotoxicity assays at a 1:1 E:T ratio. (J) Representative FACS analysis of cytotoxicity with target cells labeled with eFluor 670 (shown in blue). (K) Enzyme-linked immunosorbent assay showing high-level production of the inflammatory cytokines IL-2, IFN-γ, and TNFα by CD1a CARTs exposed to Jurkat and NALM6 (negative control) cells in 16-hour assays at a 1:1 E:T ratio (n = 4). n.s., not significant. *P < .05, **P < .01, ***P < .001.
CD1a CARTs specifically eradicate T-ALL cell lines and primary blasts in vitro
An initial analysis of CD1a density in cell surface confirmed the high expression level of the target antigen specifically in coT-ALL primary cells, primografts, and cell lines (Figure 2F-G), further validating CD1a as an immunotarget. Consequently, CD1a CARTs were first tested in vitro using the CD1a+ T-ALL cell lines Jurkat and MOLT4, and the B-ALL cell line NALM6 as a negative control. Compared with control CARTs (either MOCK T cells or CD22 CARTs), CD1a CARTs specifically eliminated CD1a+ T-ALL cells in a manner dependent on the E:T ratio. A relatively low E:T ratio of 2:1 or 4:1 induced 50% to 90% specific cell lysis in 16-hour assays (Figure 2H; supplemental Figure 2). Importantly, CD1a+ T-ALL cells barely survived exposure to CD1a CARTs in a 72-hour absolute number assay at a 1:1 E:T ratio (Figure 2I-J). CD1a CARTs produced high levels of the proinflammatory cytokines IL-2, TNFα, and IFN-γ on coculture with CD1a+ cell lines, confirming their cytotoxicity (Figure 2K).
To further address their ability to eliminate primary tumors, CD1a CARTs were cocultured with primary coT-ALL samples (either freshly harvested or in a patient-derived xenograft [PDX] model), with a proportion of CD1a+ blasts ranging between 80% and 99% (Figure 3A). Compared with MOCK T cells, CD1a CARTs specifically eliminated primary CD1a+ coT-ALL cells in 48-hour cytotoxicity assays at a 4:1 E:T ratio (Figure 3B-C). BM normal hematopoietic cells (CD1a–) as well as CD1a– T-ALL blasts were not lysed by CD1a CARTs, further confirming the specificity of the CD1a CAR (Figure 3C; supplemental Figure 5). High-levels of IFN-γ and TNFα were also secreted on coculture with CD1a+ primary T-ALL cells (Figure 3D).Collectively, CD1a CARTs have a potent and specific antileukemic activity against coT-ALL cell lines and primary blasts in vitro.
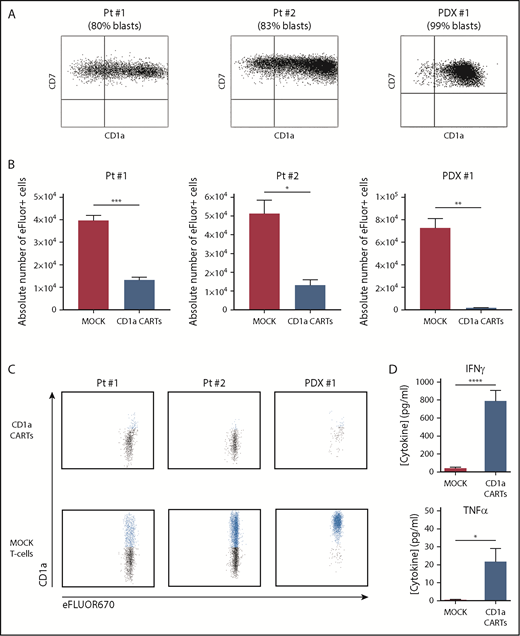
CD1a CARTs specifically target and eliminate in vitro CD1a+ T-ALL blasts from primary samples or PDX models. (A) Expression of CD1a vs CD7 in coT-ALL blasts from primary patients/primografts. The percentage of CD1a+ blasts is indicated. (B) Cytotoxicity (in absolute counts of eFluor-positive cells) measured by using FACS in 48-hour cytotoxicity assays at a 4:1 E:T ratio (n = 3). (C) Representative FACS analysis of CD1a (shown in blue) within the eFluor-labeled target cells at the end of the cytotoxicity assay, revealing specificity of CD1a CARTs (n = 3). (D) High-level production of pro-inflammatory cytokines by CD1a CARTs analyzed according to enzyme-linked immunosorbent assay (n = 3 independent supernatants) in 16-hour assays at a 4:1 E:T ratio. *P < .05, **P < .01, ***P < .001, ****P < .0001.
CD1a CARTs specifically target and eliminate in vitro CD1a+ T-ALL blasts from primary samples or PDX models. (A) Expression of CD1a vs CD7 in coT-ALL blasts from primary patients/primografts. The percentage of CD1a+ blasts is indicated. (B) Cytotoxicity (in absolute counts of eFluor-positive cells) measured by using FACS in 48-hour cytotoxicity assays at a 4:1 E:T ratio (n = 3). (C) Representative FACS analysis of CD1a (shown in blue) within the eFluor-labeled target cells at the end of the cytotoxicity assay, revealing specificity of CD1a CARTs (n = 3). (D) High-level production of pro-inflammatory cytokines by CD1a CARTs analyzed according to enzyme-linked immunosorbent assay (n = 3 independent supernatants) in 16-hour assays at a 4:1 E:T ratio. *P < .05, **P < .01, ***P < .001, ****P < .0001.
CD1a CARTs exhibit potent antileukemic activity in vivo
We next evaluated the activity of CD1a CARTs in vivo using both Luc-expressing Jurkat T-ALL cells (Figure 4; supplemental Figure 3) and a primary coT-ALL xenograft model38 (Figure 5). NSG mice were transplanted with 3 × 106 Luc-expressing Jurkat cells 3 days before IV infusion of either 2 × 106 or 5 × 106 CD1a (or MOCK) CARTs, and leukemia establishment was followed up weekly by using BLI. In contrast to the mice receiving MOCK T cells, which showed massive tumor burden by BLI, those mice given CD1a CARTs were practically leukemia free by day 25. The control of leukemia progression was CD1a CART cell dose-dependent. Flow cytometry analysis of tumor burden in PB confirmed the BLI data. Importantly, FACS analysis revealed T-cell persistence in all hematopoietic tissues analyzed; however, we found a significantly increased biodistribution of CD1a-directed effector T cells in BM and spleen, compared with the biodistribution of MOCK T cells, indicative of an active control of disseminated leukemia by CD1a CARTs.
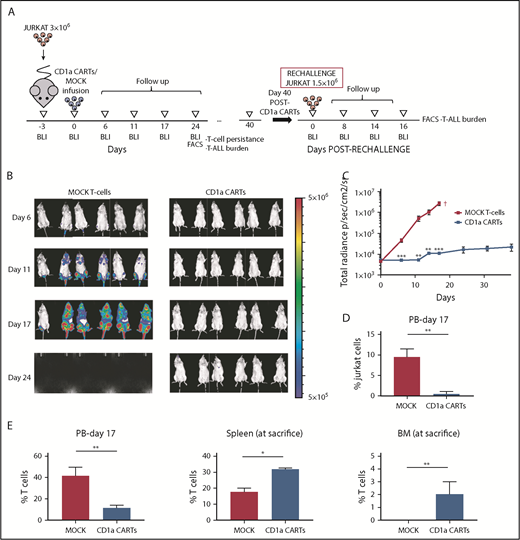
CD1a CARTs fully control the progression of coT-ALL cells in a mouse xenograft setting. (A) Scheme of the xenograft model. NSG mice (n = 6/group) were IV injected with 3 × 106 Luc/GFP–expressing Jurkat cells followed 3 days after by a single IV injection of 5 × 106 MOCK or CD1a CARTs. Tumor burden was monitored every 4 to 6 days according to BLI using IVIS imaging. When MOCK-treated animals were fully leukemic, one-half of the CD1a CART-treated animals were euthanized and analyzed by using FACS (BM, PB, and spleen) for leukemic burden and CART persistence. The remaining animals were rechallenged 6 weeks later with 1.5 × 106 Luc-Jurkat cells and were followed up as before. (B) IVIS imaging of tumor burden monitored by BLI at the indicated time points. (C) Total radiance quantification at the indicated time points. †Euthanization. (D) Circulating Jurkat cells in PB 17 days after CARTs infusion. (E) T-cell persistence in PB at day 17, and spleen and BM at euthanization. Data are shown as mean ± SD (n = 6 mice/group). *P < .05, **P < .01, ***P < .001.
CD1a CARTs fully control the progression of coT-ALL cells in a mouse xenograft setting. (A) Scheme of the xenograft model. NSG mice (n = 6/group) were IV injected with 3 × 106 Luc/GFP–expressing Jurkat cells followed 3 days after by a single IV injection of 5 × 106 MOCK or CD1a CARTs. Tumor burden was monitored every 4 to 6 days according to BLI using IVIS imaging. When MOCK-treated animals were fully leukemic, one-half of the CD1a CART-treated animals were euthanized and analyzed by using FACS (BM, PB, and spleen) for leukemic burden and CART persistence. The remaining animals were rechallenged 6 weeks later with 1.5 × 106 Luc-Jurkat cells and were followed up as before. (B) IVIS imaging of tumor burden monitored by BLI at the indicated time points. (C) Total radiance quantification at the indicated time points. †Euthanization. (D) Circulating Jurkat cells in PB 17 days after CARTs infusion. (E) T-cell persistence in PB at day 17, and spleen and BM at euthanization. Data are shown as mean ± SD (n = 6 mice/group). *P < .05, **P < .01, ***P < .001.
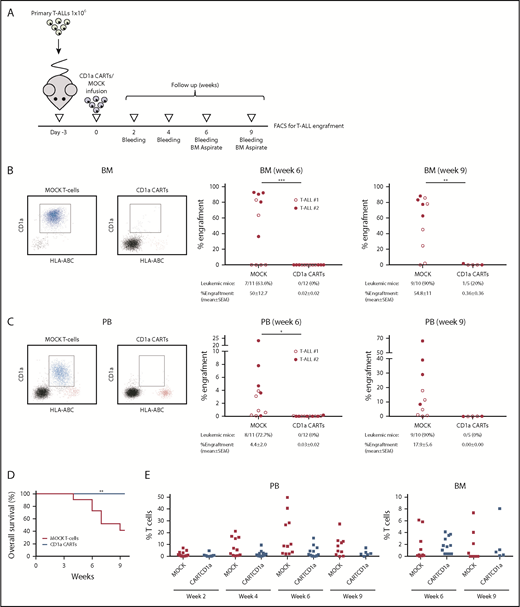
CD1a CARTs fully abolish the progression of primary CD1a+ coT-ALL blasts in a PDX setting. (A) Scheme of the PDX model. NSG mice (n = 5–6/group) were IV injected with 1 × 106 primary coT-ALL cells followed 3 days after by a single IV injection of 1 × 106 MOCK or CD1a CARTs. Tumor burden was monitored according to FACS every other week by bleeding and BM aspirate after 6 and 9 weeks. Frequency of leukemic mice and levels of leukemia in BM (B) and PB (C) 6 and 9 weeks after infusion of CARTs. The left panels show representative FACS plots. Primary CD1a+ T-ALL blasts are shown in blue, effector T cells are shown in red, and mouse cells are shown in black. (D) Nine-week OS of coT-ALL primografts receiving either CD1a CARTs or MOCK T cells. (E) Effector T-cell persistence over time in PB (week 2 toward week 9) and BM (weeks 6 and 9). Each dot represents an independent mouse. *P < .05, **P < .01, ***P < .001, Malcolm-Cox test.
CD1a CARTs fully abolish the progression of primary CD1a+ coT-ALL blasts in a PDX setting. (A) Scheme of the PDX model. NSG mice (n = 5–6/group) were IV injected with 1 × 106 primary coT-ALL cells followed 3 days after by a single IV injection of 1 × 106 MOCK or CD1a CARTs. Tumor burden was monitored according to FACS every other week by bleeding and BM aspirate after 6 and 9 weeks. Frequency of leukemic mice and levels of leukemia in BM (B) and PB (C) 6 and 9 weeks after infusion of CARTs. The left panels show representative FACS plots. Primary CD1a+ T-ALL blasts are shown in blue, effector T cells are shown in red, and mouse cells are shown in black. (D) Nine-week OS of coT-ALL primografts receiving either CD1a CARTs or MOCK T cells. (E) Effector T-cell persistence over time in PB (week 2 toward week 9) and BM (weeks 6 and 9). Each dot represents an independent mouse. *P < .05, **P < .01, ***P < .001, Malcolm-Cox test.
In a clinically more relevant PDX model of coT-ALL, NSG mice were first transplanted with 1 × 106 primary CD1a+ T-ALL blasts followed 3 days later by infusion of 1 × 106 CD1a (or MOCK) CARTs, and leukemia engraftment was then followed up biweekly by bleeding and end point BM analysis (Figure 5A). Engraftment of CD1a+ coT-ALL cells gradually increased over time, both in BM (50% ± 13% and 55% ± 11% on weeks 6 and 9, respectively) (Figure 5B) and PB (4.4% ± 2% and 18% ± 6% on weeks 6 and 9) (Figure 5C) in MOCK T-cell–treated PDXs and was associated with a significantly lower 9-week OS (42% vs 100%; P = .01) (Figure 5D). In contrast, CD1a CARTs fully abolished T-ALL growth/engraftment even 9 weeks after CART infusion (0.36% and 0% T-ALL blasts in BM and PB), and, importantly, they persisted in PB and BM over time (Figure 5E).
In vivo persistent CD1a CARTs are functional in rechallenge assays
Because the persistence of CARTs in hematopoietic tissues is a major biological parameter for their clinical success, we next assessed whether CD1a CARTs persisting after 40 to 50 days remained functional and efficient in controlling T-ALL progression. To do this, T-ALL–transplanted mice in which the leukemia was cleaned on treatment with CD1a CARTs were rechallenged with either Luc/Jurkat cells (Figure 6A-D) or primary T-ALLs from primografts (Figure 6E-G). Opposite to control mice in which the secondary leukemias rapidly (as soon as 2 weeks after) and massively engrafted, T-ALL engraftment was barely detectable according to either BLI or FACS in the Jurkat or primograft model after 6 weeks. Importantly, FACS analysis confirmed persisting effector T cells in PB, BM, and spleen of rechallenged animals, further supporting the functional effect of CD1a CARTs in controlling disease progression in rechallenge assays.
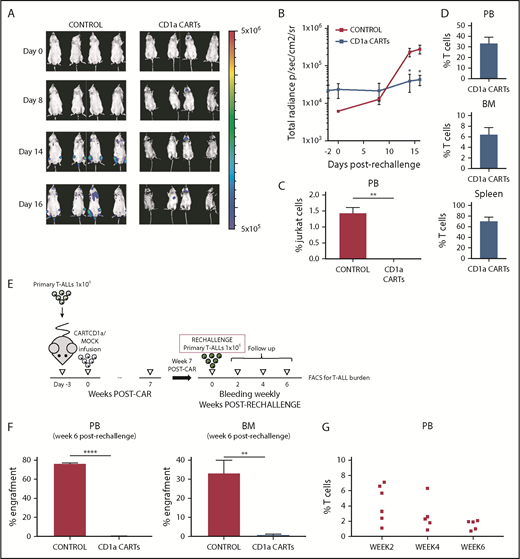
CD1a CARTs retain the ability to control progression of CD1a+ cell lines and coT-ALL primary samples in a rechallenge PDX setting. (A) IVIS imaging of Jurkat cells burden in the rechallenged mice. (B) Total radiance quantification over time in the mice rechallenged with Jurkat cells. (C) Circulating Jurkat cells in PB 16 days after rechallenge. (D) Robust effector T-cell persistence in PB, BM, and spleen at euthanization of the rechallenged animals. (E) Scheme of the rechallenge PDX experiments using coT-ALL primary samples. CART-bearing PDX mice were rechallenged with 1 × 106 primary CD1a+ T-ALL 7 weeks after initial CART infusion. (F) Secondary coT-ALL burden in engrafted PB (left panel) and BM (right panel) 6 weeks after leukemia rechallenge. (G) Effector T-cell persistence over time in PB (weeks 2, 4, and 6) from PDXs rechallenged with coT-ALL primary samples. Each dot represents an independent mouse. *P < .05, **P < .01, ****P < .0001.
CD1a CARTs retain the ability to control progression of CD1a+ cell lines and coT-ALL primary samples in a rechallenge PDX setting. (A) IVIS imaging of Jurkat cells burden in the rechallenged mice. (B) Total radiance quantification over time in the mice rechallenged with Jurkat cells. (C) Circulating Jurkat cells in PB 16 days after rechallenge. (D) Robust effector T-cell persistence in PB, BM, and spleen at euthanization of the rechallenged animals. (E) Scheme of the rechallenge PDX experiments using coT-ALL primary samples. CART-bearing PDX mice were rechallenged with 1 × 106 primary CD1a+ T-ALL 7 weeks after initial CART infusion. (F) Secondary coT-ALL burden in engrafted PB (left panel) and BM (right panel) 6 weeks after leukemia rechallenge. (G) Effector T-cell persistence over time in PB (weeks 2, 4, and 6) from PDXs rechallenged with coT-ALL primary samples. Each dot represents an independent mouse. *P < .05, **P < .01, ****P < .0001.
Patient-derived CD1a CARTs specifically target autologous CD1a+ blasts and retain antiviral activity
The proper choice of the target antigen and avoiding T-cell fratricide are crucial for the success of CARTs in T-ALL. Accordingly, we examined whether PB-derived mature CD3+CD1a– T cells from patients with coT-ALL can be isolated and genetically modified to express CD1a CAR. For this experiment, CD3+CD1a– T cells from patients were isolated (>95% purity; data not shown), activated with CD3/CD28, and lentivirally transduced (31%-70% transduction) with CD1a CAR or MOCK. We next investigated the cytolytic capacity of CD1a CARTs derived from primary T-ALLs against active T-ALL patient-matched PBMCs (Figure 7A). Total PBMCs were used as targets because this method allows assessment of both the autologous cytotoxicity potential and the degree of fratricide. Within eFluor 670–labeled target PBMCs, the great majority (∼75%) are CD1a+ blasts, and ∼15% are CD3+CD1a– mature T cells (Figure 7B). Compared with MOCK T cells, the CD1a CARTs exhibited massive and specific cytolytic capacity against autologous CD1a+ blasts but not against CD1a– mature T cells (Figure 7C), further showing that CD1a CARTs are fratricide resistant.
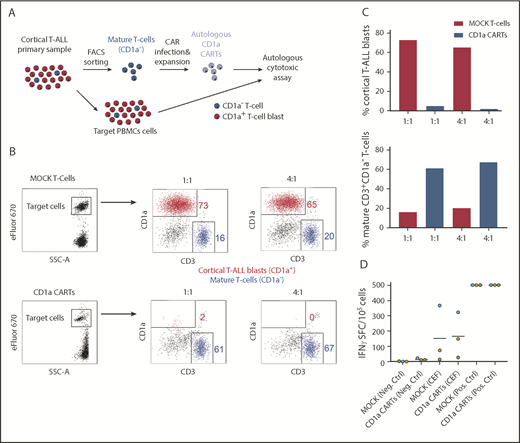
CD1a CARTs derived from patients with coT-ALL at presentation (specifically lyse autologous CD1a+ T-ALL blasts). (A) Scheme depicting the experimental design for the autologous cytotoxic assay. Mature (normal) CD3+CD1a– T cells were FACS-purified from the PB of a patient with coT-ALL, infected with CD1a CAR, expanded, and exposed to autologous total PBMCs. (B) FACS analysis of autologous cytotoxic 48-hour assay at 1:1 and 4:1 E:T ratios. eFluor 670–labeled total PBMC target population contains CD1a+ T-ALL blasts (red) and mature CD3+CD1a– T cells (blue). (C) Quantification of CD1a CART-mediated specific lysis for coT-ALL blasts (upper panel) and CD3+CD1a– mature T cells (bottom panel). (D) ELISpot showing the number of IFN-γ SFC from mock vs CD1a CARTs stimulated with a pool of peptides from CMV, EBV, and flu (CEF). Staphylococcal enterotoxin B was used as positive control. SFC, spot-forming cell.
CD1a CARTs derived from patients with coT-ALL at presentation (specifically lyse autologous CD1a+ T-ALL blasts). (A) Scheme depicting the experimental design for the autologous cytotoxic assay. Mature (normal) CD3+CD1a– T cells were FACS-purified from the PB of a patient with coT-ALL, infected with CD1a CAR, expanded, and exposed to autologous total PBMCs. (B) FACS analysis of autologous cytotoxic 48-hour assay at 1:1 and 4:1 E:T ratios. eFluor 670–labeled total PBMC target population contains CD1a+ T-ALL blasts (red) and mature CD3+CD1a– T cells (blue). (C) Quantification of CD1a CART-mediated specific lysis for coT-ALL blasts (upper panel) and CD3+CD1a– mature T cells (bottom panel). (D) ELISpot showing the number of IFN-γ SFC from mock vs CD1a CARTs stimulated with a pool of peptides from CMV, EBV, and flu (CEF). Staphylococcal enterotoxin B was used as positive control. SFC, spot-forming cell.
To further assess the potential thymic toxicity of CD1a CARTs, we next used human normal fetal thymus-derived CD7+ thymocytes as target cells. Only the CD1a+ cortical thymocytes (red) were eliminated by the CD1a CARTs, whereas developmentally earlier and later CD1a– (blue) thymic T-lineage populations (CD7+CD34+ and CD7+CD34–) were not targeted (supplemental Figure 4), limiting the on-target/off-tumor effects to a developmentally transient thymic population of cortical thymocytes. We finally sought to determine whether CD1a CARTs can protect, by themselves, the host by targeting the most common pathogens causing viremia in immunosuppressed patients. To do this, we tested the reactivity of CD1a CARTs to CMV, EBV, and flu antigens (CEF) and quantified the SFCs by INF-γ ELISpot. Both MOCK T cells and CD1a CARTs responded very similarly to stimulation with viral peptides, suggesting that CD1a CARTs retain antiviral activity (Figure 7D).
Discussion
T-ALL is an aggressive hematological cancer with poor clinical outcome, both in children and adults, for which there is currently no targeted therapy.46,47 Salvage chemotherapy regimens induce remissions in only 20% to 50% of R/R cases, and allogeneic HSPC transplantation is largely associated with toxicities.4 Despite intensive multiagent chemotherapy protocols, 5-year survival remains ∼50%,4,41 reinforcing the need for novel targeted therapies. Similarly, the only targeted therapies previously used for eradication of malignant T cells, with suboptimal clinical outcome, are the ricin A chain toxin–conjugated mAbs anti-CD5 and anti-CD7.48,49
Adoptive cellular immunotherapy based on CARs holds great promise in cancer-targeted treatment. T cells can be modified to specifically recognize and eliminate tumor cells through the expression of CARs, which redirect genetically modified T cells to specific antigen-expressing tumor cells in a major histocompatibility complex–independent manner.9,10 However, broadening the scope of CARTs to treat T-ALL and T-cell lymphomas has proven challenging because of the shared expression of target antigens between CARTs and T-lineage tumoral cells.15-20 Accordingly, 2 major stumbling blocks need to be overcome for the use of CARTs for T-ALL and T-cell lymphoma: first, CARTs self-targeting/fratricide occurs when CARTs recognize pan T-cell antigens; and second, CARTs targeting pan T-cell antigens will induce T-cell aplasia, leading to a life-threating immunodeficiency.15-19
We hypothesized that the choice of the antigen against which we wish to re-direct T cells would represent a major advance to solving the problems associated with the shared expression of T-cell markers between normal and malignant T cells. CD1a is a lipid-presenting molecule whose expression is basically restricted to coT-ALL, retained at relapse, and is practically absent in human tissues with the exception of cortical thymocytes, skin LC, and some circulating myeloid dendritic cells during development.28,29,50,51 Given this scenario, we opted for the CD1a antigen as a feasible and safe target for CAR immunotherapy in R/R coT-ALL, the most common subtype of T-ALL.21,28,52
We developed and functionally characterized CD1a-specific CARTs, which displayed robust cytotoxicity against T-ALL cell lines and primary CD1a+ coT-ALL cells, both in vitro and in vivo in xenograft models. The CD1a CARTs continuously expanded 200-fold, similar to MOCK T cells, showing that redirecting CARTs against CD1a antigen does not induce T-cell fratricide. Also, the use of CD1a CARTs for coT-ALL bypasses the need for sophisticated genome editing–based disruption of target antigens in T cells before CAR transduction as a strategy to avoid self-antigen–driven fratricide.15-17,19 We further showed that in steady-state hematopoiesis, CD1a is exclusively expressed in a subset of CD34+CD7+ cortical thymic T-progenitors, whereas earlier CD34highCD7high T-progenitors lack CD1a. In addition, neither normal CD34+ HSPCs nor mature T cells from multiple tissues express CD1a during ontogeny, thereby minimizing the risk of on-target/off-tumor toxicity. Indeed, when human fetal thymus-derived CD7+ thymocytes were exposed to CD1a CARTs, only the CD1a+ cortical thymocytes were eliminated by the CD1a CARTs. Developmentally earlier and later thymic T-lineage populations (CD34+ and CD34–) were not targeted, limiting the on-target/off-tumor effects to a developmentally transient thymic population of cortical thymocytes and further confirming the fratricide-resistant nature of CD1a CARTs.
Regarding safety, we do not expect irreversible toxicities or immunodeficiency attributed to CD1a CARTs for the following reasons: (1) CD1a+ thymocytes represent a transient and thymus-restricted population, eventually regenerated by “nontargetable” upstream CD34+CD7+CD1a– T-cell progenitors physiologically/constantly maturing into functional T cells; (2) CD1a CARTs themselves respond normally to viral antigens and therefore are likely to be protective against pathogens; (3) the clinical use of specific antibodies against CD5 or CD748 did not reveal severe or irreversible toxicities; and (4) postnatal thymectomy does not lead to immunodeficiency in humans,53,54 likely because thymic emigrants generated early in life persist for decades,53 suggesting that potential transient elimination of thymic progenitors by CD1a CARTs in pediatric patients would not compromise the complete antiviral T-cell repertoire in adult life. Nonetheless, whether infants could eventually develop a premature immunosenescence later in life merits caution, as this finding was reported for infants thymectomized before 1 year of age.55-59 Fortunately, however, T-ALL is extremely infrequent in infants. Safer ultimate strategies would include the implementation of an inducible molecular switch to control potential toxicities linked to CARTs60-62 and/or the use of CD1a CARTs as a therapy bridge before curative allogeneic HSPC transplantation.
Beyond the hematopoietic system, CD1a expression in humans is restricted to LCs in the skin, which constitute a very rare subset of dendritic cells in the epidermis.63 The immunological homeostasis of LCs is vital to avoid inflammatory skin diseases such as dermatitis and psoriasis. The in vivo role of CD1a has long remained a challenge because CD1a is not expressed in mice. However, a recent seminal paper based on mice with transgenic expression of CD1a64 showed that CD1a drives the pathogenesis of poison-induced dermatitis and psoriasis.63 Importantly, the authors also showed that treatment with CD1a-blocking antibodies alleviated skin inflammation with no comorbidity or side effects. This finding highlights that CD1a is a potential therapeutic target in inflammatory skin diseases but also supports CD1a as a safe target for R/R coT-ALL. A phase 1 clinical trial is planned in our institution to confirm the safety of CD1a-directed CARTs in R/R coT-ALL. Unfortunately, CD1a-directed CAR is not a therapeutic choice for the aggressive ETP T-ALL, which is commonly CD1a–.
Last but not least, it remains to be determined whether autologous mature T cells can be recovered from the PB of patients with T-ALL, modified to express the CAR of interest, and retain cytolytic activity against the tumoral cells expressing the target antigen. Similarly, flow cytometric analysis of PB from patients with active T-ALL revealed the presence of mature CD3+CD1a– T cells in all the patients. These PB-derived T cells were efficiently harvested from patients with coT-ALL, modified to express CD1aCAR. They exhibited a potent and specific cytolytic activity against autologous CD1a+ T-ALL blasts, reinforcing the notion that CD1a CARTs are fratricide resistant. Furthermore, as a note of safety, the genome-wide mutational landscape of both pediatric and adult T-ALL65 has revealed 106 somatic oncogenic drivers present in diagnostic samples but absent in remission mononuclear cells and T cells, suggesting that mature T cells would be a relatively safe source of autologous T cells for CAR transduction. We propose CD1a CART immunotherapy once tumor burden has been extensively reduced with standard therapies. Alternatively, donor T cells would represent an ideal effector source in patients with coT-ALL who previously underwent allogeneic HSPC transplantation. Finally, universal, “off-the-shelf” allogeneic-suitable T cells are the short-term “holy grail” for leukemia immunotherapies because T-cell lymphopenia or T-cell dysfunction occurs in many multitreated R/R patients with hematological malignances. As such, preliminary studies have already shown the potential of using universal allo-tolerant off-the-shelf CARTs generated by genomic editing–mediated deletion of receptors such as CD3, T-cell receptor, or β2-microglobulin, which are essential for antigen recognition and immune function in a major histocompatibility complex–dependent context.15,19 We provide preclinical evidence for the therapeutic and safe use of fratricide-resistant CD1a CARTs for R/R coT-ALL.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Josep Nomdedeu (Hospital Sant Pau, Barcelona) and Vicky Martínez (Hospital Virgen de la Arrixaca, Murcia) for providing diagnostic immunophenotyping data, Neus Villamor (Hospital Clinic of Barcelona, Barcelona, Spain) and Jorge Rossi and Marisa Felice (Garrahan Pediatric Hospital, Buenos Aires, Argentina) for providing FACS and clinical data, and Julio Castaño, Maria Castellá, and Paolo Petazzi (members of Pablo Menéndez laboratory) for fruitful technical discussions. They are also indebted to Maksim Mamonkin (Baylor College of Medicine, Houston, TX) for helping them with T-cell expansion.
This research was supported by the European Research Council (H2020) (CoG-2014-646903), the Agencia Estatal de Investigación/European Regional Development Fund (SAF2016-80481-R and SAF2016-75442-R), and the Catalunya Government (SGR330 and PERIS 2017) (P.M.), as well as the Asociación Española Contra el Cáncer, Beca FERO, and the ISCIII/FEDER (PI17/01028) (C.B.). P.M. also acknowledges institutional support from the Obra Social La Caixa-Fundaciò Josep Carreras. J.G.P. holds a Miguel Servet contract (CP15/00014), and O.B.-L. is supported by an AGAUR-FI fellowship from the Catalan Government. P.M. is an investigator of the Spanish Cell Therapy cooperative network (TERCEL).
Authorship
Contribution: D.S.-M. conceived the study, designed and performed experiments, analyzed data, and wrote the paper; M.L.B., F.G.-A., H.R.-H., M.T., J.J., T.V.-H., O.B.-L., J.G.P., B.U., and J. Calvo performed experiments and analyzed data; M.C., J.L.F., M.R.-O., S.G.-G., F.P., J. Cools, and M.L.T. provided primary T-ALL samples, primografts, and FACS data; C.B. performed experiments and financially supported the study; and P.M. conceived the study, designed experiments, wrote the manuscript, and financially supported the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pablo Menéndez, Josep Carreras Leukaemia Research Institute, School of Medicine, Barcelona University, Carrer Casanova 143, 08036 Barcelona, Spain; e-mail; pmenendez@carrerasresearch.org; and Diego Sánchez-Martínez, Josep Carreras Leukaemia Research Institute, School of Medicine, Barcelona University, Carrer Casanova 143, 08036 Barcelona, Spain; e-mail: dsanchez@carrerasresearch.org.
