Abstract
Mutations in the cytosolic 5′ nucleotidase II (NT5C2) gene drive resistance to thiopurine chemotherapy in relapsed acute lymphoblastic leukemia (ALL). Mechanistically, NT5C2 mutant proteins have increased nucleotidase activity as a result of altered activating and autoregulatory switch-off mechanisms. Leukemias with NT5C2 mutations are chemoresistant to 6-mercaptopurine yet show impaired proliferation and self-renewal. Direct targeting of NT5C2 or inhibition of compensatory pathways active in NT5C2 mutant cells may antagonize the emergence of NT5C2 mutant clones driving resistance and relapse in ALL.
Introduction
Pediatric ALL represents the most prominent example of the success of combination chemotherapy in the treatment of cancer, with cure rates >80%.1 However, the prognosis for patients at relapse remains poor.2 Analyses of paired diagnostic and relapsed acute lymphoblastic leukemia (ALL) samples support a role for Darwinian selection of relapse-associated mutations driving resistance to cytotoxic agents and glucocorticoids during disease progression.3-9
Early in the development of chemotherapy in the 1950s, therapy with methotrexate and 6-mercaptopurine (6-MP) induced transient hematologic remissions in childhood leukemia.10,11 As multiagent chemotherapy protocols developed, this concept was integrated in the postremission treatment of ALL in the form of 6-MP and methotrexate maintenance therapy, resulting in markedly reduced relapsed rates and improved long-term remissions.12 Today, thiopurine-based maintenance therapy is universally included in the treatment of ALL, and patients with optimal dose-intensity and compliance have improved rates of event-free survival.13,14 Consistently, ALL relapse is frequently associated with 6-MP resistance, most commonly as result of gain-of-function mutations in the cytosolic 5′ nucleotidase II (NT5C2) gene.3-6 NT5C2 mutations are found in 3% to 10% of relapsed early precursor B-cell ALL cases and 20% of relapsed T-cell ALL cases and are particularly common among early-relapse patients, accounting for 35% to 45% of these cases.3-6 NT5C2 mutations can also be found in acute promyelocytic leukemia relapse samples from patients who have been treated with 6-MP.15 In addition, PRPS1 mutations encoding hyperactive PRPS1 proteins promoting enhanced purine biosynthesis have also been associated with thiopurine resistance.16 Furthermore, loss of MSH6 can induce 6-MP resistance via impaired DNA mismatch repair complex signaling of thioguanine nucleotide DNA lesions.17,18 Here, we review the mechanisms of NT5C2 mutations as drivers of resistance to therapy and the potential role of mutant NT5C2 as a therapeutic target in relapsed ALL.
NT5C2 mutations in relapsed leukemia
A role for increased breakdown of 6-MP–derived toxic nucleotides in leukemia resistance to 6-MP therapy was first hypothesized in 1988 by Pieters and Veerman,19 who subsequently found that some leukemias with high cytosolic 5′ nucleotidase activity showed in vitro resistance to 6-thiopurine treatment.20 However, the driver role of cytotoxic thiopurine nucleotide inactivation in 6-MP resistance was only formally established 25 years later with the identification of NT5C2 activating mutations in relapsed ALL.3-6
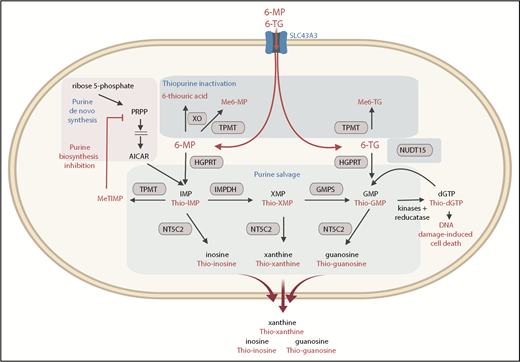
NT5C2 (EC3.1.3.5, cN-II, high Km 5′-nucleotidase) is an evolutionarily conserved and ubiquitously expressed nucleotidase enzyme that preferentially mediates the 5′-dephosphorylation of the 6-hydroxypurine monophosphates inosine monophosphate (IMP), guanosine monophosphate (GMP), and xanthine monophosphate (XMP), as well as the deoxyribose forms of IMP and GMP. Physiologically, NT5C2 counterbalances the activity of nucleoside kinases and regulates the purine nucleotide pool by facilitating the export of the resulting purine nucleosides out of the cell.21-28 Additionally, NT5C2 can also dephosphorylate the thiopurine monophosphate nucleotides resulting from the incorporation of 6-MP and 6-thioguanine into the salvage pathway of purine biosynthesis (Figure 1).29

Purine and thiopurine metabolism. NT5C2 dephosphorylates endogenous and thiopurine-generated 6-hydroxypurine monophosphates before they are exported out of the cell. AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; dGTP, deoxyguanosine triphosphate; GMPS, guanosine monophosphate synthase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; IMPDH, inosine monophosphate dehydrogenase; Me6-MP, methyl 6-MP; Me6-TG, methyl 6-thioguanine; MeTIMP, methyl thioinosine monophosphate; NUDT15, Nudix hydrolase 15; PRPP, phosphoribosyl pyrophosphate; TPMT, thiopurine methyltransferase; XO, xanthine oxidase.
Purine and thiopurine metabolism. NT5C2 dephosphorylates endogenous and thiopurine-generated 6-hydroxypurine monophosphates before they are exported out of the cell. AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; dGTP, deoxyguanosine triphosphate; GMPS, guanosine monophosphate synthase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; IMPDH, inosine monophosphate dehydrogenase; Me6-MP, methyl 6-MP; Me6-TG, methyl 6-thioguanine; MeTIMP, methyl thioinosine monophosphate; NUDT15, Nudix hydrolase 15; PRPP, phosphoribosyl pyrophosphate; TPMT, thiopurine methyltransferase; XO, xanthine oxidase.
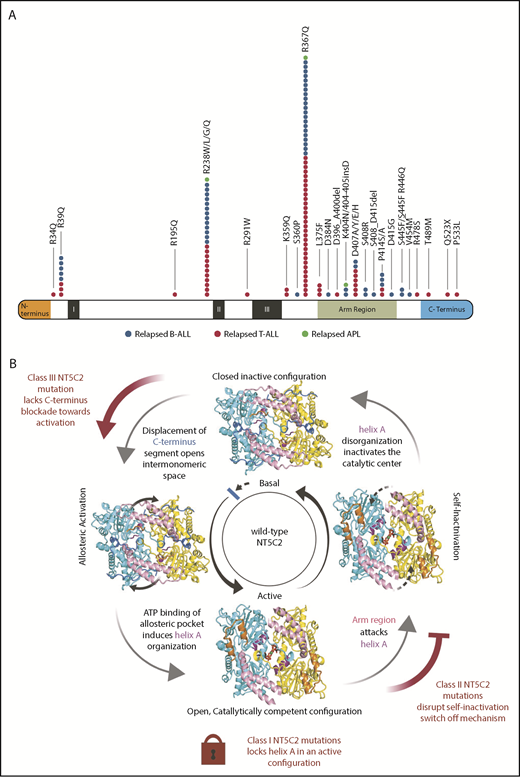
To date, 32 independent NT5C2 mutant alleles have been described, consisting of 27 single amino acid substitutions, 4 in-frame indel mutations, and a C-terminal truncating mutation.3-7,15,30-32 NT5C2 point mutations commonly involve specific residues (Arg39, Arg238, Arg367, Leu375, Asp407, and Pro414) and are frequently recurrent, with NT5C2 R367Q standing out as the most common relapse-associated NT5C2 mutation, accounting for 90% of mutant cases. Consistently with the presence of NT5C2 heterozygous mutations, mutational hotspots, and common recurrent amino acid substitutions suggestive of a gain-of-function mechanism of action (Figure 2A),33,34 analysis of relapse-associated NT5C2 mutant proteins showed increased enzymatic activity in nucleotidase assays.3,6

NT5C2 relapse-associated mutations. (A) NT5C2 relapse-associated mutations in B-cell ALL, T-cell ALL, and acute promyelocytic leukemia (APL). (B) Schematic representation of NT5C2 regulation. NT5C2 dimer protein is depicted with the arm region shown in pink, C terminus in royal blue, N terminus in orange, and helix A in purple. The arm region shown in gray represents a prediction based on modeling. Class I NT5C2 mutants lock the protein in an active helix A configuration. Class II NT5C2 mutants disrupt a switch-off mechanism mediated by the arm and intermonomeric pocket regions. The class III NT5C2 mutant protein lacks the C-terminus stabilizing element, leaving the protein in a more open configuration.
NT5C2 relapse-associated mutations. (A) NT5C2 relapse-associated mutations in B-cell ALL, T-cell ALL, and acute promyelocytic leukemia (APL). (B) Schematic representation of NT5C2 regulation. NT5C2 dimer protein is depicted with the arm region shown in pink, C terminus in royal blue, N terminus in orange, and helix A in purple. The arm region shown in gray represents a prediction based on modeling. Class I NT5C2 mutants lock the protein in an active helix A configuration. Class II NT5C2 mutants disrupt a switch-off mechanism mediated by the arm and intermonomeric pocket regions. The class III NT5C2 mutant protein lacks the C-terminus stabilizing element, leaving the protein in a more open configuration.
Interestingly, rs72846714, an intronic single-nucleotide polymorphism in the NT5C2 locus is associated with the propensity for thioguanine DNA incorporation in ALL patients.35 Although rs72846714-A is not associated with increased ALL relapse risk, it has been linked with increased occurrence of gain-of-function NT5C2 relapse-specific mutations, supporting a potential interaction between somatic and germline variants in driving increased NT5C2 activity.35
Mechanisms of NT5C2 activation
Structurally, NT5C2 is a member of the haloacid dehalogenase superfamily of Mg2+-dependent intracellular 5′-nucleotidases36 and forms a homotetramer composed of a dimer of dimers.21,37 Mechanistically, each dimer of NT5C2 represents a functional unit competent for catalysis and is regulated by allosteric activators including adenosine triphosphate, deoxyadenosine triphosphate, and Ap4A21,38-40 (Figure 2B), which induce reconfiguration of a segment known as the helix A region (Gly355-Glu364) from a disordered state in the apo inactive form of the enzyme to an ordered α-helix conformation. This ordering of the helix A segment into an α-helix configuration activates the enzyme by opening and reconfiguring the catalytic center of NT5C2, making it competent for substrate binding and catalysis.37,41
Crystallization, structural modeling, and biochemical characterization of NT5C2 mutant proteins have defined 3 classes of relapse-associated NT5C2 mutants with different mechanisms of action (Figure 2B). Class I NT5C2 mutations (NT5C2 K359Q and L375F) force the helix A segment into an α-helix active configuration, resulting in high levels of nucleotidase activity in the absence of allosteric activators.30 In contrast, class II NT5C2 mutant proteins show increased nucleotidase activity in basal conditions but still dynamically respond to allosteric activation.30 Mechanistically, class II mutants disrupt positively charged residues located in the intermonomeric pocket (NT5C2 R39Q, R238W/L/G/Q, R367Q, S445F_R446Q, and R478S) or alter the arm region (K404N, 404-405insD, D407A/Y/E/H, S408R, P414S/A, and D415G), disrupting a bipartite regulatory switch responsible for NT5C2 self-inactivation.30 This switch-off mechanism is mediated by the arm region, and the intermonomeric space is triggered by the opening of the otherwise tightly closed NT5C2 dimer following NT5C2 activation.30 This conformational change turns the intermonomeric space between the two NT5C2 subunits into a positively charged cavity, which is now invaded by Asp407, a negatively charged residue located in the tip segment of the flexible arm region of NT5C2 (Glu373-Tyr434). At the bottom of the intermonomeric positively charged cavity, Asp407 interacts with and destabilizes the active helix A, turning off the enzyme. Finally, NT5C2 class III mutations are defined by a C-terminal truncating allele (NT5C2 Q523*), a mutant whose nucleotidase activity in basal conditions is similar to that of the wild-type NT5C2 protein but shows a markedly increased response to allosteric activation.30 This mutation removes the C-terminal tail of NT5C2 consisting of a stretch of negatively charged residues, which by interacting with the positive charges along the intermonomeric surface of the NT5C2 dimer secures a tightly closed protein conformation in basal conditions.30 Mechanistically, removal of the C-terminal tail of NT5C2 by the Q523* mutation induces a more open conformation and enhances the response to allosteric activators.30
In heterozygous NT5C2 mutant cells, homotypic (mutant/mutant or wild-type/wild-type) and heterotypic (wild-type/mutant) dimers can be combined in different tetramer configurations (wild-type/mutant: 4/0, 3/1, 2/2, 1/3, 0/4). In the case of the wild-type/R367Q dimers the increased nucleotidase activity seems to be mediated by the catalytic center of the mutant subunit.42 In contrast, in wild type/R238W dimers, increased nucleotidase activity requires an active catalytic center in the neighboring wild-type subunit.42
NT5C2 mutations confer resistance to 6-MP
The presence of gain-of-function NT5C2 mutations in relapsed leukemias following treatment with 6-MP and their association with early relapse under 6-MP therapy supports a role for NT5C2 mutations in 6-MP resistance. Mechanistically, increased NT5C2 activity can enhance the dephosphorylation and clearance of nucleotide monophosphates, 6-thioinosine monophosphate, 6-thioxanthine monophosphate, and 6-thioguanosine monophosphate, generated by 6-MP, and thus reduces the subsequent generation of methyl thio-IMP and deoxythioguanosine triphosphate, which mediate the cytotoxic effects of 6-MP via inhibition of de novo purine biosynthesis and activation of postreplicative DNA-mismatch–induced apoptosis, respectively (Figure 1).43 Consistently, expression of relapse-associated NT5C2 mutations induced selective resistance to thiopurine chemotherapy in ALL cell lines,3,6 and relapsed ALL xenografts harboring the NT5C2 R367Q mutation showed resistance to 6-MP.4
Dynamics of clonal selection and therapy resistance
A characteristic feature of NT5C2 mutations in ALL is their specific association with relapse. This is in contrast with other resistance-associated genetic alterations, which can be found present both at diagnosis and at relapse in some cases and just at the time of relapse in others. Moreover, the presence of multiple subclonal NT5C2 mutations in some cases at relapse5 and clonal evolution mapping analyses support that NT5C2 mutations may occur as late events in the clonal evolution of the disease, complementing the effect of preexisting relapse driving mutations, to allow minimal residual disease progression under 6-MP chemotherapy. In agreement, analysis of isogenic Nt5c2 wild-type and Nt5c2 R367Q mutant mouse ALL tumors generated from a conditional knockin mouse model with inducible expression of the Nt5c2 mutation demonstrated acquisition of 6-MP resistance upon expression of the Nt5c2 R367Q allele and positive selection of Nt5c2 R367Q-expressing cells upon treatment with 6-MP in vitro and in vivo. However, in the absence of 6-MP, expression of Nt5c2 R367Q in this model resulted in partially impaired tumor cell growth in vitro, delayed leukemia progression in vivo, and markedly reduced leukemia-initiating cell activity.4 These loss-of-fitness phenotypes can result from aberrantly increased Nt5c2 activity, leading to dephosphorylation of IMP, XMP, and GMP and subsequent increased export of the resulting nucleosides from the cell. In the absence of 6-MP treatment, NT5C2 gain-of-function mutations are associated with a loss-of-fitness phenotype and would be outcompeted by wild-type cells. In contrast, the presence of an activating NT5C2 mutant allele in minimal residual cells would result in resistance to 6-MP therapy, positive selection, and clonal expansion, leading to progression under therapy and relapse. Deep sequencing with unique molecular identifier barcodes and ultrasensitive droplet polymerase chain reaction analyses failed to identify NT5C2 mutations in diagnostic samples from patients who subsequently relapsed with NT5C2 mutant leukemia, with a sensitivity of 1:1000 and 1:20 000, respectively, supporting that NT5C2 mutations may not be present at the time at diagnosis but are acquired in the context of minimal residual disease and selected for during maintenance therapy. Consistently, analysis of serial samples during remission can detect the emergence and expansion of NT5C2 mutations several weeks before the occurrence of relapse.4
Targeting NT5C2 mutant relapsed ALL
In vitro, resistance to 6-MP in the context of NT5C2 mutations can be overcome by increased dose intensity; however, this approach may not be feasible to implement in the clinic because of 6-MP dose-limiting toxicities (myelosuppression). Combination therapy with 6-MP and an NT5C2 inhibitor is predicted to block the selection of NT5C2 mutations as a genetic path toward relapse. NT5C2 inhibitors are under development,44-47 and these efforts will surely be enhanced by the availability of high-resolution crystal structures of mutant NT5C2. Although a mutant-selective inhibitor would offer maximum specificity, structure analyses of NT5C2 wild-type and mutant proteins show little differences in the catalytic center, questioning the feasibility of developing mutant specific NT5C2 inhibitors. Of note, homozygous loss-of-function mutations in NT5C2 cause congenital autosomal recessive spastic paraplegia-45 (OMIM 613162), a neurological syndrome characterized by lower limb spasticity and mental retardation,48 setting a cautionary note regarding potential neurological toxicities of an NT5C2 inhibitor. However, it is possible that inhibition of wild-type NT5C2 may still be tolerated. In this context, although class I mutations may require direct targeting of the catalytic center, class II and III NT5C2 mutant proteins retain sensitivity to allosteric regulation, suggesting that small-molecule inhibitors targeting the allosteric site may effectively abrogate their capacity to confer resistance to 6-MP.
Finally, excess export of purine nucleosides in cells harboring relapse-associated NT5C2 mutations is predicted to enhance purine biosynthesis via activation of allosteric feedback loops. Partial compensation of purine nucleotide depletion by increased synthesis renders these tumors more sensitive to inhibition of IMP dehydrogenase, a central enzyme mediating the conversion of IMP to XMP downstream of both the de novo and salvage pathways of purine biosynthesis, offering a potential alternative therapeutic strategy toward abrogation of NT5C2 mutant clones.4
Conclusions
Activating mutations in NT5C2 are specifically associated with relapsed leukemias following treatment with 6-MP. These genetic alterations can deplete intracellular nucleotide pools, imposing a fitness cost to leukemia lymphoblasts, yet they also confer resistance to 6-MP, resulting in their positive selection during maintenance treatment as drivers of progression under therapy. Targeting collateral vulnerabilities and the combination of NT5C2 inhibitors with 6-MP may preclude the emergence of NT5C2 mutant leukemia clones, cutting down the occurrence of relapse.
Acknowledgments
This work was supported by Translational Research grant 6531-18 and Screen To Lead grant 8011-18 from the Leukemia & Lymphoma Society (A.F.), National Institutes of Health, National Cancer Institute grants CA206501 and CA210065 (A.F.), an Innovative Research Award from the Alex Lemonade Stand Foundation (A.F.), and the Hyundai Hope On Wheels Scholar Award (A.F.). C.L.D. was supported by National Institutes of Health, National Cancer Institute training grant T32-CA09503.
Authorship
Contribution: C.L.D. and A.F. wrote the manuscript
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Adolfo Ferrando, 1130 St. Nicholas Ave, ICRC 402A, New York, NY 10032; e-mail: af2196@columbia.edu.