

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory syndrome induced by aberrantly activated macrophages and cytotoxic T cells. The primary (genetic) form, caused by mutations affecting lymphocyte cytotoxicity and immune regulation, is most common in children, whereas the secondary (acquired) form is most frequent in adults. Secondary HLH is commonly triggered by infections or malignancies but may also be induced by autoinflammatory/autoimmune disorders, in which case it is called macrophage activation syndrome (MAS; or MAS-HLH). Most information on the diagnosis and treatment of HLH comes from the pediatric literature. Although helpful in some adult cases, this raises several challenges. For example, the HLH-2004 diagnostic criteria developed for children are commonly applied but are not validated for adults. Another challenge in HLH diagnosis is that patients may present with a phenotype indistinguishable from sepsis or multiple organ dysfunction syndrome. Treatment algorithms targeting hyperinflammation are frequently based on pediatric protocols, such as HLH-94 and HLH-2004, which may result in overtreatment and unnecessary toxicity in adults. Therefore, dose reductions, individualized tailoring of treatment duration, and an age-dependent modified diagnostic approach are to be considered. Here, we present expert opinions derived from an interdisciplinary working group on adult HLH, sponsored by the Histiocyte Society, to facilitate knowledge transfer between physicians caring for pediatric and adult patients with HLH, with the aim to improve the outcome for adult patients affected by HLH.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) has become more widely recognized in adults, with all ages affected. Patients often suffer from recurrent fever, cytopenia, liver dysfunction, and a sepsis-like syndrome that may rapidly progress to terminal multiple organ failure. Subspecialists in hematology/oncology, infectious diseases, rheumatology/clinical immunology, gastroenterology/hepatology, neurology, emergency medicine, intensive care, and general medicine are challenged by this rare multifaceted syndrome. Physicians should be aware of HLH, because early recognition may prevent irreversible organ damage and subsequent death.1,2
Although familial (primary) HLH (FHL), a major HLH subtype in children, can also occur in adolescents and young adults, secondary (acquired) HLH (sHLH) is by far the most common in these age groups. The treatment protocols HLH-94 and HLH-2004 have been established as scientific cornerstones for diagnosis, classification, and treatment of HLH in patients younger than 18 years.3-5
Our current views on HLH are driven by lessons learned in pediatrics, and pediatricians still often consult on adults with HLH. However, HLH triggers, organ reserve, fitness, and clinical presentation differ between the pediatric and adult age groups. Transferring pediatric precepts regarding pathogenesis, diagnostics, and treatment of HLH to adult patients may confer risks. Therefore, the HLH Steering Committee of the Histiocyte Society developed these recommendations for diagnosis and treatment of HLH in adults, as a complement to previously published recommendations on etoposide-based therapy in HLH.6
Methods
Procedure
The recommendations are based on expert opinion supported by best available evidence from studies supporting the individual statements. They were initially proposed and discussed by e-mail and then selected and structured in a telephone conference. After further refinement by e-mail, each statement was discussed, revised, and voted on in face-to-face meeting, followed by final refinement by e-mail. The consensus strength for the statements was classified as follows:
Strong consensus: >95% of participants agree
Consensus: 75% to 95% of participants agree
Majority agreement: 50% to 75% of participants agree
No consensus: ≤50% of participants agree
Constitution of Recommendation Committee
The authors represent the current members of the Working Group “HLH in Adults” of the Histiocyte Society (www.histiocytesociety.org). The current head of the Working Group (P.L.R.) served as coordinator. The following medical subspecialties were represented: adult hematology/oncology and internal medicine (P.L.R., R.M., N.B., S.B., M.G., Y.W., Z.W.), pediatric hematology/oncology (T.v.B.G., M.B.J., A.K., K.E.N., G.J., J.-I.H.), rheumatology/clinical immunology (adult: J.A.M.v.L., A.V.R; pediatric: A.H.), intensive care (adult: G.L.; pediatric: M.H.), and genetics/clinical immunology (J.G.-H.).
Pathogenesis and epidemiology of HLH
Statement 1: Primary and secondary HLH, including MAS-HLH, are hyperferritinemic hyperinflammatory syndromes with a common terminal pathway but with different pathogenetic roots (strong consensus).
HLH is an aberrant hyperinflammatory hyperferritinemic immune response syndrome that is driven by T cells and associated with a potentially fatal cytokine storm.7,8 The term “macrophage activation syndrome” (MAS; or MAS-HLH) refers to a subset of patients with HLH arising on a background of systemic autoinflammation/autoimmunity and should be restricted to patients with Still’s disease, lupus, vasculitis, and other related autoimmune systemic diseases, because its treatment may differ from that recommended for other forms of sHLH9,10 (see Statement 14).
The current view of sHLH pathogenesis is an inability of the immune system to adequately restrict stimulatory effects of various triggers.7,10,11 Inherited variations in HLH-associated genes, which are well characterized in pediatric HLH, may play a role in adult-onset HLH, but acquired immune dysfunction in response to infections, malignancies, and autoinflammatory/autoimmune disorders seems to be the leading cause in adults (Table 1).10,12,13
Statement 2: HLH triggers, including occult malignancies, require a meticulous search for the underlying disease that should be continued, despite ongoing HLH treatment (strong consensus).
Infections are the most prevalent triggers of HLH.10,14 In adults, particularly with increasing age, malignancies, primarily lymphomas, are another major cause (Table 1).10,15-17 A variety of malignancies are associated with HLH in adults, including T-cell or natural killer (NK) cell lymphomas (35%), B-cell lymphomas (32%), leukemias (6%), Hodgkin lymphoma (6%), other hematologic neoplasms (14%), solid tumors (3%), and other nonspecified neoplasms (3%).10,15
The epidemiology of HLH varies substantially as a result of population heterogeneity and variable underlying triggers.17-22 A large literature review on adult HLH reported a mean age at HLH onset of 49 years (63% males).10 The reported incidence of malignancy-associated HLH varies from 1% in patients with hematological malignancies (0.36/100 000 individuals per year)17 to a cumulative incidence rate of 2.8% in patients with malignant lymphoma23 and 9% of patients with acute myeloid leukemia (AML) after intensive induction therapy.22
Diagnosis of HLH in adults
Statement 3: The diagnosis of HLH in adults should be based on the HLH-2004 diagnostic criteria in conjunction with clinical judgment and the patient’s history (strong consensus).
The HLH-2004 diagnostic criteria
In 1991, the Histiocyte Society proposed a standardized set of 5 diagnostic criteria for HLH used for the prospective HLH-94 clinical trial.24 These criteria were revised for HLH-2004: individuals needed to meet ≥5 of 8 diagnostic criteria (Table 2).4,5 On occasion, HLH may be strongly considered, and HLH-directed therapy may be initiated, even though 5 criteria are not fulfilled (see Statements 4, 6, 7, 10, and 13).6
Hyperferritinemia should always prompt inclusion of HLH in the differential diagnosis.25 Ferritin values characteristic of HLH in adults are often >7000 to 10 000 µg/L and, rarely, may be >100 000 μg/L.26 Ferritin levels >10 000 µg/L are >90% sensitive and specific for HLH in children, although other criteria need to be met to make the diagnosis. Although a ferritin level in this range should also raise a strong suspicion of HLH in adults, hyperferritinemia is less specific. Integration of a number of clinical features is required to confirm the diagnosis HLH in adults.27,28 Soluble interleukin-2 (IL-2) receptor (also soluble CD25 [sCD25]), 1 of the diagnostic criteria in HLH-2004, has recently been reported as a good to excellent low-cost diagnostic test for adult HLH, with an area under the curve of 0.90 (95% confidence interval, 0.83-0.97) compared with an area under the curve of 0.78 (95% confidence interval, 0.67-0.88) for ferritin.29,30 The HLH-2004 criteria, developed for children, are not validated formally for adults and remain based on expert opinion. Various case series have used modified HLH-2004 criteria.19,21,31-41
Other diagnostic tools
Other features supporting an HLH diagnosis that are not part of the HLH-2004 criteria include hyperbilirubinemia, hepatomegaly, transaminitis (present in the vast majority of patients with HLH), and elevated lactate dehydrogenase and d-dimer levels, with the latter usually elevated even when international normalized ratio, partial thromboplastin time, and fibrinogen are normal. These findings may help to discriminate HLH from septic shock and conditions such as autoimmune hemolytic anemia, and they are also useful in assessing response to therapy.
The HLH-probability calculator (HScore), with graded clinical and laboratory parameters, is a Web-based online calculator (http://saintantoine.aphp.fr/score/) developed retrospectively in adult patients that may be a helpful diagnostic tool (Table 3).42 The pattern of inflammatory cytokines (elevated levels of interferon-γ [IFN-γ] and IL-10, with only modestly elevated IL-6 levels) has high diagnostic accuracy for sHLH and may be a useful approach to differentiate HLH from infection and to monitor patients; however, the utility of this pattern of changes needs to be verified in children and adults outside of China.43-45 For diagnosis using functional assessment of cytotoxicity and genetics, see Statement 4.
Clinical phenotypes
Many adult patients with HLH present with the triad of fever, bicytopenia with potential bleeding diathesis, and splenomegaly. Wasting and fatigue may occur. Edema, purpura, dyspnea, diarrhea, diffuse bleeding, icterus, and an overall sepsis-like appearance are extremes of severe HLH with onset of organ failure. Mild initial signs, including recurrent fevers, lymphadenopathy, organomegaly, rash, and arthralgias, may progress with an unexpected rapidity and severity. These signs and symptoms, despite adequate antimicrobial therapy and/or without detectable infectious focus, along with a dramatic clinical progression serve as red flags for possible HLH. No single clinical or laboratory parameter has sensitivity and specificity to allow an unambiguous HLH diagnosis. Close clinical observation with repeated physical examinations and laboratory assessments are mandatory for diagnosis.
Statement 4: Diagnostic tests for genetic HLH include functional assessment of lymphocyte cytotoxicity and guided genetic testing. They are useful for detecting potential genetic predisposition to HLH in select patients, but pending results must not delay the clinical decision to treat HLH (consensus).
Functional and genetic testing are not generally recommended in adult patients with HLH because abnormalities are rarely detected. Genetic defects leading to FHL, with impairment of lymphocyte cytotoxicity, and genetic alterations in related disorders are presented in Table 4.13,46-50 Although most patients with underlying genetic defects show the features summarized in Table 4, cellular expression of perforin, SLAM-associated protein, X-linked inhibitor of apoptosis protein proteins, or CD107a (a glycoprotein necessary for degranulation [exocytosis] of perforin-containing granules from cytotoxic lymphocytes) may be normal or nearly normal in rare cases.50 To determine whether a patient has an inherited form of HLH, genetic analyses (ie, next-generation sequencing, gene panels, whole-exome sequencing), along with functional testing (ie, NK cell cytotoxicity and CD107a upregulation), will be needed.10,15-17,46,51 This is particularly true for patients with a family history of consanguinity and/or HLH, partial albinism, or recurrent disease and for young male adults with Epstein-Barr virus (EBV) infection or lymphoproliferation. Testing should also be considered for HLH patients with unknown trigger. Importantly, pending results must not delay the clinical decision to treat HLH.
Although primary HLH is rare in adults, mutations in HLH-associated genes can be detected in adult patients with HLH (Table 5)51 ; in the United States, a rate of 7% (12/175) is reported.52 However, the predominant locus, the A91V mutation in PRF1, is considered a hypomorphic mutation that is present in up to 10% of healthy whites.53 Although these hypomorphic alleles may constitute a risk factor for HLH, the vast majority of individuals carrying these mutations have no clinical symptoms.
Statement 5: Lymphoma as a hidden trigger of HLH may be difficult to detect. Use of positron emission tomography–guided imaging, repetitive tissue sampling, and consultation with a lymphoma reference pathologist, are recommended (strong consensus).
About 40% to 70% of HLH cases in adults are malignancy associated, triggered by the malignancy itself at presentation or seen after initiation of chemotherapy; the latter may even occur in patients in remission and is thought to be due to immunosuppression and/or infection.15 Therefore, patients need a thorough cancer workup, with special consideration of Hodgkin lymphoma and non-Hodgkin lymphoma (NHL). Atypical presentation and certain lymphoma subtypes (ie, Hodgkin; diffuse large B-cell lymphoma, intranasal subtype; NK/T-cell lymphoma; angioimmunoblastic T-cell lymphoma; anaplastic large cell lymphoma; panniculitis-like T-cell NHL; intravascular B-cell lymphoma; and peripheral T-cell lymphoma) are more strongly associated with HLH.54,55 Computed tomography scan enhanced by positron emission tomography, followed by biopsy of suspicious lesions, is advised to reveal occult disease.56 An elevated sCD25/ferritin ratio has been reported in lymphoma-associated HLH.57
Tumor-infiltrating reactive lymphocytes can mask an underlying lymphoma. Thus, a close interaction among clinicians, pathologists, and immunologists is required to determine the correct diagnosis. In individuals with HLH of unknown cause and splenomegaly, splenectomy may be considered to detect lymphomas hiding in the spleen or perisplenic tissue. This is justified in clinical circumstances that strongly support lymphoma (history of B symptoms, weight loss, elevated sCD25/ferritin ratio).58
Treatment of adult patients with HLH
Statement 6: HLH-94 treatment components, including etoposide, are highly effective in treating hyperinflammation in adults with HLH (Figure 1) (strong consensus).
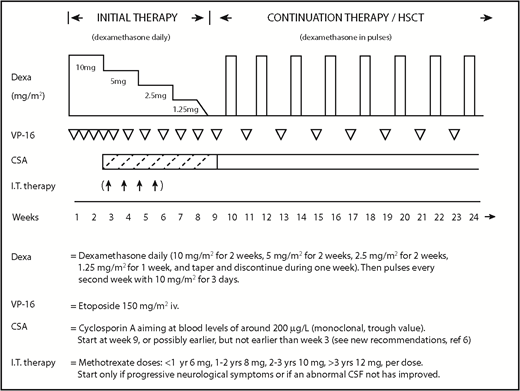
Overview of the HLH-94 treatment protocol. Note that dose and frequency adjustments of this protocol are advised for adult patients (see Statement 7).
Overview of the HLH-94 treatment protocol. Note that dose and frequency adjustments of this protocol are advised for adult patients (see Statement 7).
The heterogeneity of adult HLH prohibits a “1-size-fits-all protocol” (Figure 2). The HLH-94 protocol drastically improved the nearly uniformly fatal outcome in pediatric HLH to a long-term survival >50%.3 The HLH-94 protocol consists of corticosteroids, typically dexamethasone, cyclosporine A (CSA), intrathecal therapy, and etoposide, to delete activated T cells and suppress inflammatory cytokine production (Figure 1).6 Etoposide, a chemotherapeutic agent, has high specificity against T-cell proliferation and cytokine secretion in mice.59 However, adult, and especially, elderly patients may have chronic comorbidities, making them more vulnerable to end organ damage caused by cytokine storm in HLH and HLH-94 chemotherapy. A reduced etoposide frequency, from twice weekly to once a week, with or without a reduction in dose from 150 mg/m2 to 50-100 mg/m2, should be considered. In the HLH-2004 study, CSA was administered upfront instead of after 8 weeks, as in HLH-94, and pre–stem cell transplantation (SCT) mortality was reduced from 27% to 19% (P = .064 adjusted for age and sex). Because this improvement was not significant and because CSA is associated with side effects, as well as contraindications, particularly early in the disease course, HLH-94 remains the recommended standard of care.5 In HLH-94, intrathecal therapy is only suggested in case of progressive neurological symptoms after 2 weeks of therapy or if an abnormal cerebrospinal fluid has not improved by then (Figure 1).
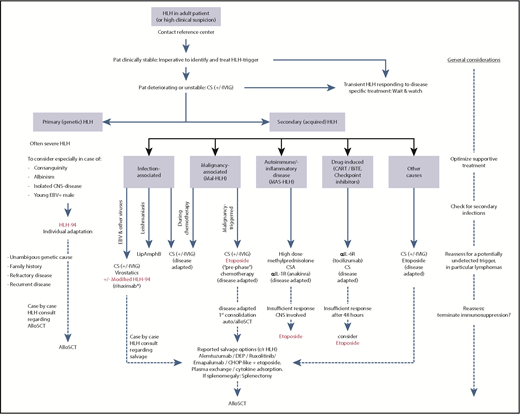
Treatment algorithm for adult patients with HLH, including MAS-HLH. The HLH-94 pediatric treatment protocol is the consensus mainstay treatment of newborns, toddlers, and children up to 18 years of age, where genetic causes of HLH are enriched. Individual adaptation regarding the length and dosing of the HLH-94 treatment plan in adults is warranted. Allogeneic hematopoietic stem cell transplant (alloSCT) can cure primary HLH and may be applied in patients with high-risk hematologic malignancy as consolidation treatment or in relapsed HLH after successful salvage treatment. Treatment in adults cannot be standardized and needs tailoring according to the underlying condition and HLH-initiating trigger (infection, malignancy, autoimmune/autoinflammatory, drug induced, other causes). In relapsed/refractory (r/r) HLH, treatment intensification with chemotherapy, use of the anti-CD52 antibody alemtuzumab, cytokine adsorption using filter columns or plasma exchange, off-label treatment with the JAK2 inhibitor ruxolitinib, or the anti–IFN-γ antibody emapalumab have shown reasonable efficacy. BiTE, bispecific T-cell engager; CART, chimeric antigen receptor T cells; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CS, corticosteroids; DEP, doxorubicin, etoposide, methylprednisolone; IVIG, polyvalent immunoglobulins; Pat, patient. *Off-label in EBV-HLH.
Treatment algorithm for adult patients with HLH, including MAS-HLH. The HLH-94 pediatric treatment protocol is the consensus mainstay treatment of newborns, toddlers, and children up to 18 years of age, where genetic causes of HLH are enriched. Individual adaptation regarding the length and dosing of the HLH-94 treatment plan in adults is warranted. Allogeneic hematopoietic stem cell transplant (alloSCT) can cure primary HLH and may be applied in patients with high-risk hematologic malignancy as consolidation treatment or in relapsed HLH after successful salvage treatment. Treatment in adults cannot be standardized and needs tailoring according to the underlying condition and HLH-initiating trigger (infection, malignancy, autoimmune/autoinflammatory, drug induced, other causes). In relapsed/refractory (r/r) HLH, treatment intensification with chemotherapy, use of the anti-CD52 antibody alemtuzumab, cytokine adsorption using filter columns or plasma exchange, off-label treatment with the JAK2 inhibitor ruxolitinib, or the anti–IFN-γ antibody emapalumab have shown reasonable efficacy. BiTE, bispecific T-cell engager; CART, chimeric antigen receptor T cells; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CS, corticosteroids; DEP, doxorubicin, etoposide, methylprednisolone; IVIG, polyvalent immunoglobulins; Pat, patient. *Off-label in EBV-HLH.
The risk of children developing treatment-related AML in the HLH-2004 and HLH-94 studies was 0.3% (1/368) to 0.4% (1/249) at a median follow-up of 5.2 and 6.2 years, respectively,3,5,6 and 1.2% (1/81) in patients with EBV-associated HLH (EBV-HLH) treated with a median cumulative etoposide dose of 1500 mg/m2 body surface area, with a median follow-up of 44 months.60 The need to stay below a cumulative dose of 2-3 g/m2 should be kept in mind, particularly in HLH patients without malignancy.60
Allogeneic SCT
Adults with primary HLH may need allogeneic hematopoietic SCT (alloSCT), which has dramatically improved outcomes in children.61 Transplant-related mortality in children using reduced-intensity conditioning (RIC) has been reported to compare favorably to myeloablative conditioning (MAC).62 A retrospective European Society of Blood and Marrow Transplantation study did not show superiority of RIC over MAC in adults.63 Decisions regarding transplantation should be made on clinical grounds and in consultation with experts in HLH and alloSCT. Inactive HLH before transplantation is strongly associated with better survival.64 Patients with primary HLH and nonmalignant sHLH may be candidates for RIC, as well as MAC. The pretransplant conditioning regimen in malignancy-associated HLH (Mal-HLH) may be guided by disease-specific protocols, as provided by local standards, which includes MAC to optimally control the underlying disease. In patients in whom HLH-causing mutations are detected, HLA typing of close relatives should also integrate screening for the same gene mutations, to avoid a stem cell source with the same pathogenic biallelic mutation(s).
Statement 7: The variable severity of HLH, including MAS-HLH, demands graded intensity and length of treatment. Treatment should be tailored to control hyperinflammation and to treat identified disease triggers (strong consensus).
An HLH diagnosis may be suspected, but not confirmed, by the presence of 5 of 8 HLH-2004 criteria. Resolution of HLH without HLH-specific treatment has been observed, particularly in infection-associated HLH.65 In moderately active HLH, the decision to start HLH-directed treatment depends on clinical judgment and assessment of organ function. Pulsed corticosteroids and elements of HLH-94 (dexamethasone 10 mg/m2 with/without a modified dose of etoposide) may be used. A clear indication for immediate administration of etoposide is severe HLH presenting with imminent organ failure.33,66 Individualized modified HLH-94–like treatment has been suggested in sHLH.67 Because etoposide is primarily cleared by the kidneys, dose reduction is recommended if renal function is impaired based on age-specific norms (for dose recommendations see Ehl et al6 ), whereas no dose reduction of etoposide is recommended for isolated hyperbilirubinemia and/or elevated transaminases.
Addition of IV immunoglobulin (IVIG) (up to 1.6 g/kg in split doses over 2-3 days) may be considered,9 because IVIG has anti-inflammatory potential by inhibiting complement activation, blocking antibody Fc fragments and macrophage Fc receptors, and neutralizing cytokines.68,69 However, the use of IVIG has been questioned, at least in adult-onset Still’s disease.70 Anakinra may reduce mortality in sepsis patients with MAS features.71
Many patients with sHLH require <8 weeks of etoposide. Although patients may be continued on the full course (8 weeks) of etoposide in the absence of major toxicities, we recommend a weekly reevaluation of the need for continued etoposide therapy. Patients with residual disease after 8 weeks may benefit from maintenance therapy and, possibly, alloSCT. For those requiring alloSCT because of underlying genetic mutations, HLH-94 maintenance therapy is often recommended after the initial 8 weeks of therapy.6 CSA may be replaced by tacrolimus, but both need careful drug level monitoring and toxicity assessment.27
Statement 8: Secondary infections are a major cause of fatality and may erroneously be diagnosed as HLH relapse (strong consensus).
Certain HLH triggers carry the inherent risk of acquired cellular immunodeficiency, such as Hodgkin lymphoma, T-cell lymphomas, or HIV. Additionally, HLH-directed treatment depletes leukocytes (T/B cells and granulocytes). For patients requiring such treatment, administration of broad antimicrobial prophylaxis against Pneumocystis jirovecii and fungi is recommended. Hospitalization in units with high efficiency particulate air–filtered air should be considered. We also suggest antiviral prophylaxis because of the severe T-cell depletion.
Mal-HLH
Statement 9: Mal-HLH comes in 2 forms: “malignancy-triggered HLH” as a presenting feature of the malignancy at diagnosis or at relapse and “HLH during chemotherapy,” in most cases induced by infections. Differentiating these HLH subtypes is important, because the therapeutic approach differs markedly (strong consensus).
Malignancy-triggered HLH
Treatment of malignancy-triggered HLH needs to balance HLH-specific and tumor-specific treatment. Corticosteroids are often used as first-line treatment to combat inflammation. In highly active HLH, or if severe organ damage is imminent, dose-adjusted etoposide (50-100 mg/m2) may be used prior to tumor-specific treatment.16 Etoposide can be added to CHOP or CHOP-like protocols (cyclophosphamide, doxorubicin, vincristine, etoposide, prednisone [CHOEP] or dose-adapted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin [DA-EPOCH]).9 Patients with aggressive lymphomas should be considered for evaluation of involvement of the central nervous system (CNS), with cerebral magnetic resonance imaging and lumbar puncture.72 Prophylactic or therapeutic age-adapted high-dose IV methotrexate may be considered individually to prevent CNS relapse (this treatment is not well tolerated by older patients). Patients in remission who are eligible for treatment intensification may be candidates for autologous SCT, using high-dose etoposide-containing chemotherapy as primary consolidation; this recommendation is based on the assumption that the dismal prognosis of lymphoma-associated HLH can be overcome by high-dose chemotherapy, which has been demonstrated in T-cell NHL.73 A decision toward consolidation treatment by using alloSCT requires careful individual assessment (see Statement 12).74
The co-occurrence of HLH and lymphoma, particularly EBV-driven lymphoma in younger patients, should trigger expert consultation and germline genetic testing, because evidence of HLH-associated mutations may support primary allogeneic, and not autologous, SCT.75 Early HLA typing and donor search in Mal-HLH is advised in selected malignancies with potential indication for primary consolidation by alloSCT (eg, Burkitt lymphoma, acute leukemias, myelodysplastic syndromes).27
HLH during chemotherapy
“HLH during chemotherapy” develops during or after treatment of a malignant disorder and is probably underrecognized. In neutropenic patients after induction chemotherapy for AML, as many as 9% may develop HLH with infections (fungal, bacterial, central-line associated) as the most frequent trigger.22 Diagnosis is obscured by preexisting neutropenia, liver function abnormalities that may be attributed to toxic drug effects, and increased ferritin that may be secondary to transfusion-related iron overload.
HLH has to be considered when cytopenia is unduly prolonged after chemotherapy, fever persists in spite of antibiotic treatment, and other HLH parameters are present. These patients benefit from anti-inflammatory treatment with corticosteroids (prednisolone 1-2 mg/kg or dexamethasone 5-10 mg/m2) and, possibly, IVIG 1.6 g/kg over 2 to 3 days. Etoposide should be used sparingly, because bone marrow recovery is central for immune reconstitution. Ongoing monitoring is required to detect recurrent malignant disease as a potential alternate HLH trigger. Where available, ferritin, sCD25, and bone marrow assessment (activated macrophages and/or hemophagocytosis) can help to distinguish those who are neutropenic as the result of chemotherapy from those who have underlying HLH.22
Infection-associated HLH
Statement 10: Viral infections, in particular EBV, HIV, cytomegalovirus, or influenza, are common triggers of HLH (strong consensus).
EBV-HLH
In 1 review of 2197 adults with HLH, viral infections were the most frequent trigger, dominated by EBV.10 The prognosis for EBV-HLH has improved greatly when treated promptly using HLH-94 protocols, but the variable severity of EBV-HLH demands graded intensity and length of treatment.76 Rapid clinical deterioration, in particular in treatment-naive EBV-infected patients, mandates etoposide treatment without delay.33 A more conservative approach with a short course of corticosteroids (with/without IVIG) is justified in patients with less severe disease or improving clinical manifestations. Monitoring of ferritin, sCD25, cell counts, and EBV DNA in affected patients aids in assessing treatment response.77 EBV DNA levels >103 copies per milliliter have been reported to be relevant for the development of EBV-HLH.18
Because EBV replicates in B cells, the addition of rituximab (eg, 375 mg/m2 once weekly, 2-4 times) to HLH-directed therapy may be effective in clearing the reservoir of virus in EBV-triggered HLH.78 Monitoring of ferritin, sCD25, cell counts, and EBV DNA may guide the number of rituximab doses. However, in many cases, EBV-HLH is associated with infection of T cells and/or NK cells, irrespective of the patients’ ethnic background or the clinical course,79 so rituximab cannot replace the anti–T-cell therapy with corticosteroids with/without etoposide, as suggested above. In patients with continuously increasing or sustained high levels of EBV DNA, SCT should be considered, such as in chronic active EBV.80,81
HIV-HLH
The prognosis of HLH in patients with HIV has improved in the era of highly active antiretroviral treatment.82 Lymphomas and opportunistic infections are the most important triggers to look for. A short transient treatment of overt inflammation by corticosteroids (with/without IVIG) is recommended. In a large series of patients with HLH secondary to HIV (HIV-HLH), etoposide was administered in about half.82 The wide spectrum of potential viral triggers mandates virus-specific treatment on a case-by-case basis.
Statement 11: HLH induced by intracellular infections, such as tuberculosis, leishmaniasis, or rickettsial disease, usually does not need HLH-94–like treatment but responds to specific antimicrobial treatment (strong consensus).
Patients infected by pathogens that target the monocyte–macrophage system may develop HLH, but immunosuppression as imparted by the HLH-94 protocol should be avoided, because they usually respond well to specific antimicrobial treatment. Leishmania is an endemic pathogen around the world, and treatment with (liposomal) amphotericin B cures leishmaniasis.83 Rickettsial disease is treated by tetracyclines or chloramphenicol, whereas tuberculosis requires quadruple antibiotic treatment and adaptation according to resistance testing.84
Salvage treatment of relapsed and refractory HLH
Statement 12: Salvage treatment of adults with refractory/relapsing HLH usually requires intensification using combined chemotherapy and consolidation with alloSCT (strong consensus).
Mortality in adult HLH ranges from 20% to 88%, primarily as the result of refractory HLH, secondary infections, and progression of the underlying triggering disease.19 In a prospective study, liposomal doxorubicin, etoposide, and high-dose methylprednisolone resulted in complete remission in 27% of patients and partial remission in 49% of patients within 4 weeks.85
In infection-associated HLH, in particular EBV-HLH, reactivations are common if treatment intensity is only moderate or tapered too fast; such reactivations commonly respond to treatment intensification. For patients with EBV-HLH and persistent high EBV genome copy numbers or chronic active EBV and refractory/relapsing lymphoma, alloSCT is recommended.64,80,81,86 In primary HLH, reactivations and/or persistence of hyperinflammation are frequent until curative alloSCT is performed.5,6,61
Data on the efficacy of salvage agents for HLH are limited. The anti-CD52 antibody alemtuzumab (median dose 1 mg/kg split over a median of 4 days) has been reported to be beneficial in refractory pediatric patients; a reduced dose or prolonged maintenance (as in chronic lymphocytic leukemia) has been used in young adults.87,88 Other salvage options include CHOP-like protocols plus etoposide and targeted inhibition of JAK signaling with ruxolitinib.89,90 Plasmapheresis or the use of cytokine adsorption columns may aid in rescuing critically ill patients from a deleterious cytokine storm.91,92 In November of 2018, the U.S. Food and Drug Administration approved emapalumab (anti–IFN-γ monoclonal antibody) as a second-line therapeutic agent for primary HLH in children and adults, after completion of an initial study that included 27 patients with relapsed/refractory disease. Emapalumab has the potential to be more readily tolerated than etoposide, although there has not yet been significant experience in the treatment of adults.93
As a general rule, in patients with refractory HLH, treatment decisions need to be individualized according to the most likely triggering cause. In patients with Mal-HLH, treatment of the malignancy guides the salvage approach (intensification of chemotherapy). Contact with a HLH reference center is recommended.
HLH and MAS-HLH in the intensive care unit
Statement 13: In critically ill patients with persistent fever, cytopenias, and organomegaly, particularly in confirmed or presumed cases of sepsis, sepsis-like syndromes, and/or evolving multiorgan failure, suspicion for HLH should be raised and further HLH testing should be initiated (strong consensus).
Intensivists may be the first to diagnose HLH in patients with multiple organ dysfunction syndrome (MODS).94,95 Familiarity with HLH is important because of its nonspecific symptoms and laboratory findings, as well as that fact that the hyperinflammatory state of HLH can be observed in sepsis, MODS, and other cytokine storm syndromes. “Hyperinflammatory sepsis” (also described as “MAS-like”) refers to patients who typically have less severe hyperinflammation and may not fulfill the diagnosis of HLH.71,96-98 Importantly, HLH, MODS, and sepsis can coexist, with sepsis serving as the possible HLH trigger.2,96,99 HLH should be considered in deteriorating critically ill patients with a disproportionate inflammatory response (eg, persistent fever, unresponsiveness to vasopressors, need for extracorporeal life support), inexplicable cytopenias, and organ failure not responding to appropriate therapy, antimicrobial treatment, and aggressive supportive care.94,100 Screening for HLH should follow the HLH-2004 criteria, including bone marrow investigation (see Statement 3). Fever, 1 of the criteria, needs special consideration because it can be masked by frequent use of antipyretics, continuous renal replacement therapy, and extracorporeal life support.
Therapy should be individualized and include HLH or MAS-HLH–directed treatment (see Statements 6, 7, 10, 12, and 14), with consideration of standard supportive care and adjunctive critical care therapies.94,100-103 Reevaluation of the clinical condition should be frequent (at least every 12 hours) to determine whether initial or additional HLH-directed therapy is needed.
Clinical management of MAS-HLH
Statement 14: Treatment of MAS-HLH is different from that recommended for HLH as a result of partial pathogenetic diversity (strong consensus).
HLH in patients with underlying rheumatic conditions is historically called macrophage-activation syndrome (MAS; MAS-HLH). MAS-HLH, a form of sHLH that is increasingly recognized in adults, has been reported in association with almost all systemic rheumatic conditions.104,105 An overwhelming immune activation leads to a systemic cytokine storm, but the initiating factors might be different in MAS-HLH compared with other forms of HLH, although, like other forms of sHLH, MAS-HLH is commonly triggered by infections.8,10
Currently, there are no accepted classification criteria for adult MAS-HLH. Several criteria have been developed for children, but these still need validation in adult patients (see Statement 3).42,106-109 Notably, symptoms of MAS-HLH may be different in patients treated with biologic agents.110
Early recognition and diagnosis of MAS-HLH are essential for efficacious management. A personalized and graded treatment approach is advised.102 Conventionally, corticosteroids are the first-line treatment. High-dose pulse methylprednisolone (1 g/d for 3-5 consecutive days) is 1 frequent initial approach.111 CSA (2-7 mg/kg per day) can be added in patients with an insufficient immediate response, as well as IL-1–blocking therapy with anakinra112-114 ; a dose of 2 to 6 mg/kg up to 10 mg/kg per day subcutaneously in divided doses is suggested. Experience with anti–IL-6 blockade with tocilizumab is also increasing.114 Finally, in patients with severe active disease or CNS involvement, despite steroids, CSA, and/or anakinra, a reduced dose of etoposide (50-100 mg/m2 once weekly) may be very effective104 ; preferably, such treatment should be discussed with an expert but still not delayed.
HLH-like cytokine storm induced by novel immunotherapies
Statement 15: Novel immunotherapies may induce a cytokine storm resembling HLH that requires specific treatment (strong consensus).
With the advent of novel T-cell–engaging immunotherapies, reports of treatment-associated cytokine release syndrome have repeatedly emerged.115,116 These T-cell immunotherapies include engineered T cells, such as chimeric antigen receptor modified T (CART) cells targeting CD19, and blinatumomab, a bispecific T-cell engager antibody that connects CD3+ T cells to CD19+ target B cells.117,118 Both of these agents are approved for treatment of B-acute lymphoblastic leukemia. CART cells are also approved in relapsed/refractory B-cell NHL. CART cells and blinatumomab induce a cytokine response that strongly resembles other forms of HLH.
The anti–IL-6 antibody tocilizumab has been used with notable rapid resolution of cytokine release syndrome in patients after CART cell or blinatumomab treatment. More recently, there are increasing reports of treatment-induced HLH also in patients treated with CTLA4- and PD-1/PD-L1–directed checkpoint antibodies (ipilimumab, pembrolizumab, nivolumab, avelumab, atezolizumab), which are used in a variety of cancer subtypes. Treatment interruption or corticosteroids alone have been used with meaningful responses.119,120 A CART cell–associated toxicity working group suggests that suspected HLH should be managed with anti–IL-6 therapy and corticosteroids for organ toxicities ≥ grade 3; if the patient has no improvement clinically or serologically within 48 hours, additional therapy with etoposide (75 to 100 mg/m2) should be considered.121
Summary and perspectives
In recent years, interest in adult HLH has increased markedly; as a result, HLH is more frequently diagnosed in adults. The dramatic therapeutic success in pediatric HLH has also positively affected the survival of adults with HLH. However, there are profound differences between adult and pediatric HLH; genetic HLH is rare in adults, pediatric diagnostic criteria are suboptimal, frequent (often occult) underlying malignancies or other conditions require a different diagnostic workup, and pediatric treatment regimens may have to be adapted on a case-by-case basis.
In adults, HLH-associated mortality remains high, especially in patients with underlying malignancies. Although the drugs used in pediatric HLH are effective in adult HLH, there is a need for novel agents. Interesting trials testing alternative therapeutic approaches have been initiated, including those incorporating ruxolitinib (JAK1/2 inhibitor; ClinicalTrials.gov identifiers NCT02400463, NCT03795909, NCT03533790), anakinra (IL-1 blockade; NCT02780583), alemtuzumab (NCT02472054), and emapalumab (anti–IFN-γ monoclonal antibody; NCT01818492). It is anticipated that the increased awareness of HLH, together with a more rapid diagnostic workup and new therapeutic approaches, will improve the prognosis of HLH in adults, as has been the case in children.
Acknowledgments
This work was supported by grants from Europe for Thuringia (Thüringer Aufbaubank; P.L.R.) and from the Swedish Children's Cancer Foundation, the Swedish Cancer Foundation, the Cancer and Allergy Foundation of Sweden, and the Stockholm County Council (ALF project) (J.-I.H.).
Authorship
Contribution: P.L.R. initiated the report, which was planned and coordinated by P.L.R. and J.-I.H.; A.H., M.H., T.v.B.G., R.M., N.B., S.B., J.G.-H., M.G., M.B.J., A.K., J.A.M.v.L., G.L., K.E.N., A.V.R., Y.W., Z.W., and G.J. contributed text proposals and revisions; all authors voted on all statements; and P.L.R. and J.-I.H. drafted the manuscript, which was reviewed and approved by all authors.
Conflict-of-interest disclosure: A.H. has received speakers’ fees/honoraria from Sobi and Novartis. M.B.J. and A.K. have acted as consultants for Novimmune and Sobi. K.E.N. has received research support from Incyte Pharmaceuticals and Alpine Biosciences. A.V.R. has received speakers’ fees/honoraria from SOBI, Novartis, Lilly, and Roche. The remaining authors declare no competing financial interests.
Correspondence: Paul La Rosée, Klinik für Innere Medizin II, Hämatologie, Onkologie, Immunologie, Infektiologie und Palliativmedizin, Schwarzwald-Baar-Klinikum gGmbH, Klinikstr. 11, D-78052 Villingen-Schwenningen, Germany; e-mail: paul.larosee@sbk-vsde; and Jan-Inge Henter, Childhood Cancer Research Unit, Karolinska Institutet, Tomtebodavägen 18A, SE-171 77 Stockholm, Sweden; e-mail: jan-inge.henter@ki.se.

