

In this issue of Blood, 1 and ,2 find that homozygous germline founder mutations in genes encoding nucleotide excision repair proteins confer risk for leukemias characterized by TP53 mutations (see figure).
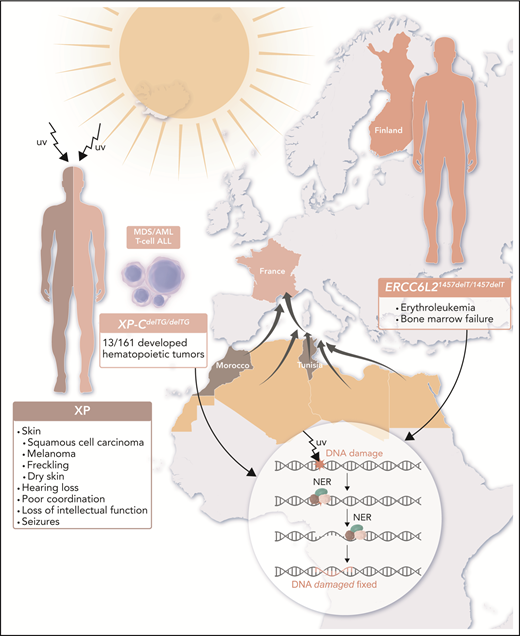
Rare patient populations in France and Finland reveal that gene mutations resulting in defective NER predispose to hematopoietic malignancies. Patients with XP are extremely sensitive to UV radiation from the sun and develop skin cancers and other problems as outlined. Homozygous founder mutations in genes encoding NER components lead to genetic susceptibility to hematopoietic malignancies: XP-CdelTG in descendants of North African immigrants to France and ERCC6L21457delT within the Finnish population. XP-C is the first NER protein to bind the damaged DNA, beginning a cascade of reactions resulting in an excised DNA fragment, repair of the gap, and resolution of the initial lesion, as shown in the inset. ERCC6L2 is part of transcription-coupled repair. When these critical NER components are absent (as is the case for homozygous XP-CdelTG mutations) or truncated prematurely (as is the case for homozygous ERCC6L21457delT mutations), TP53 mutations occur and drive leukemia development. AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; T-cell ALL, T-cell acute lymphoblastic leukemia. Professional illustration by Luk Cox, Somersault18:24.
Rare patient populations in France and Finland reveal that gene mutations resulting in defective NER predispose to hematopoietic malignancies. Patients with XP are extremely sensitive to UV radiation from the sun and develop skin cancers and other problems as outlined. Homozygous founder mutations in genes encoding NER components lead to genetic susceptibility to hematopoietic malignancies: XP-CdelTG in descendants of North African immigrants to France and ERCC6L21457delT within the Finnish population. XP-C is the first NER protein to bind the damaged DNA, beginning a cascade of reactions resulting in an excised DNA fragment, repair of the gap, and resolution of the initial lesion, as shown in the inset. ERCC6L2 is part of transcription-coupled repair. When these critical NER components are absent (as is the case for homozygous XP-CdelTG mutations) or truncated prematurely (as is the case for homozygous ERCC6L21457delT mutations), TP53 mutations occur and drive leukemia development. AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; T-cell ALL, T-cell acute lymphoblastic leukemia. Professional illustration by Luk Cox, Somersault18:24.
Defects in DNA damage repair have long been known to confer risk for human cancers. For example, Lynch syndrome derives from gene mutations in genes encoding DNA mismatch repair enzymes,3 and individuals with hereditary breast and ovarian syndrome have mutations in BRCA1/2 and other genes encoding associated DNA repair proteins. BRCA1/2 are known within our field as Fanconi anemia–like genes, whose function is essential in the bone marrow.4-6 Other cancer syndromes arise from defects in nucleotide excision repair (NER),7 including xeroderma pigmentosum (XP), which consists of 8 autosomal recessive forms (XP-A, XP-B, XP-C, XP-D, XP-E, XP-F, XP-G, and XP-V), each having a mutation in a component of the NER complex (XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, and POLH, respectively). People with XP have an extreme sensitivity to UV light, experiencing severe sunburns with minutes of exposure, dry skin (xeroderma), freckling (pigmentosum), hearing loss, poor coordination, loss of intellectual function, seizures, and development of squamous cell carcinomas and melanomas often as early as 10 years old in sun-exposed areas.
Soulier and colleagues have been following a unique cohort of 142 consanguineous families from Northern Africa, among whom 161 individuals are homozygous for a founder XP-CdelTG mutation, which causes complete absence of the XPC protein.1 Among these homozygous XP-CdelTG mutation carriers, these authors have identified 13 (8%) with myeloid malignancies and T-cell acute lymphoblastic leukemia in people aged 7 to 29 years, a frequency several-thousand–fold greater than expected in France. The myelodysplastic syndromes/acute myeloid leukemias that developed in these individuals commonly had complex karyotypes, and TP53 mutations were seen in all 5 cases for which somatic mutation data were available. Given that myeloid malignancies have not been described commonly in XP patients with other mutations and this founder mutation appears to be the only shared mutation among these individuals, the authors surmise that this susceptibility to hematopoietic malignancies is particular to this particular founder mutation.
The Finnish population also contains a founder mutation of a gene encoding a component of the transcription-coupled NER pathway, ERCC6L21457delT. This gene had previously been identified as one mutated as a germline allele in bone marrow failure syndromes.8-10 Wartiovaara-Kautto and colleagues identified 8 individuals with homozygous ERCC6L21457delT mutations who developed erythroleukemias with TP53 mutations or bone marrow failure.2
Combined, these papers present a unifying theme: mutations in NER lead to TP53 mutations, which drive the development of hematopoietic malignancies. One might expect that any gene mutation that results in problems with maintaining DNA integrity will read out as a leukemia-predisposing gene, given the tremendous replicative output of the bone marrow. Many inherited syndromes have been defined through clinical observations, but with molecular diagnostics, many of these syndromes are now recognized as encompassing more cancers than first defined. Witness expansion in understanding of BRCA1/2, originally described as mutated in hereditary breast/ovarian cancer syndrome but now understood to confer risk for a variety of solid tumors, including gastric, pancreatic, and prostate cancers and now also hematopoietic malignancies. By extension, one could imagine that we will be able to identify inherited mutations for each of the genes encoding component of DNA repair pathways, with accumulating evidence as presented here.1,2
Conflict-of-interest disclosure: The author declares no competing financial interests.


