

TO THE EDITOR:
ERCC excision repair 6 like 2 (ERCC6L2) is a newly identified gene with an impact on hematological disease development. Lack of ERCC6L2 results in defects in the transcription-coupled nucleotide excision repair pathway, leading to genome instability.1 It also affects mitochondrial function, increasing reactive oxygen species levels and altering cellular homeostasis.2
Biallelic germ line mutations in ERCC6L2 were recently reported to cause bone marrow failure (BMF).2-6 The first article described two consanguineous families where affected children experienced developmental delay and microcephaly in addition to BMF.2 However, subsequent studies have excluded these extrahematopoietic manifestations from the disease phenotype.1,3,4 Järviaho et al3 reported the ERCC6L2 c.1457delT, p.Ile486ThrfsTer36 mutation (NM_001010895.2, GRCh37; rs768081343) in 2 Finnish BMF cases.
Most BMF syndromes predispose to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Four cases of MDS and/or AML have been reported among 24 patients with biallelic ERCC6L2 mutations.1,4 These patients were diagnosed with MDS or AML in childhood or as young adults (age 2-22 years), with only 1 patient alive at the time of the original reports.1,4 AML/MDS subtypes were not reported; however, all patients were described as carrying monosomy 7.1,4 This is a common abnormality in therapy-related, secondary, erythroid, and germ line predisposed leukemias.7,8
The definition of AML with erythroid characteristics (AML M6 by French-American-British [FAB] classification) has been under debate.9,10 In practice, AML M6a and AML M6b are considered as MDS or AML, NOS, nonerythroid subtype and AML, NOS, erythroid leukemia (pure erythroid type), respectively, in the current World Health Organization (WHO) classification of myeloid malignancies.11 For clarity, we use FAB nomenclature here.
We report causality of a germ line homozygous ERCC6L2 c.1457delT, p.Ile486ThrfsTer36 mutation (supplemental Figure 1, available on the Blood Web site) resulting in early somatic TP53 mutations and AML with erythroid characteristics resembling AML M6.
Initially, we discovered 3 families with a homozygous germ line ERCC6L2 c.1457delT mutation and validated the result in a series of AML M6 patients (n = 7; excluding AML M6 arising after chemotherapy or radiation treatment for another malignancy) identified in the Finnish Hematology Registry (Figure 1B). A series of AMLs of other subtypes with whole-exome sequencing data available (n = 165) were used as a control set (Figure 1C). In the discovery families, we identified 6 individuals with the homozygous mutation (Figure 1A), all of whom had AML M6 (n = 3) or BMF (n = 3; Table 1). Only patients with BMF were alive. Additionally, 1 individual (1459, family 2) whose BM morphology data but not tissue or DNA samples were available had died as a result of AML M6.
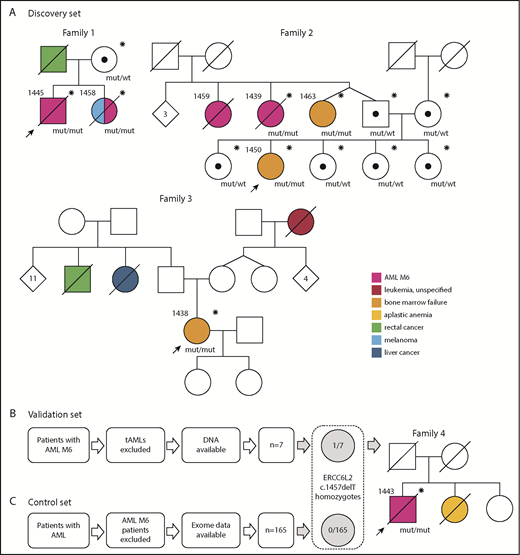
Families in this study and the selection process of patient data. Individuals who have been genotyped for the ERCC6L2 c.1457delT mutation are marked with an asterisk (*). Black dots (•) represent heterozygous carriers of the mutation. Probands in each family are marked with arrows. (A) Families (1-3) in the discovery set. Family 1: patient 1445 was age 38 years when referred to hematologist because of pancytopenia. His BM was dysplastic with strong erythroid predominance and an excess of myeloid blasts. Aiming at allogeneic hematopoietic stem cell transplantation (HSCT), his 36-year-old sister (patient 1458) was examined as a donor candidate. Tests revealed peripheral blood cytopenias. The following BM examination revealed MDS, which quickly progressed to AML M6. She died as a result of refractory leukemia. Patient 1445 underwent HSCT from a registry donor but relapsed quickly with a therapy-resistant AML M6 and died as a result of the disease. Family 2: the index patient (1450) age 18 years was diagnosed with BMF of unknown origin and referred to the hematology department in 2018. Her 2 paternal aunts (patients 1459 and 1439) had died as a result of AML M6. The twin sister (patient 1463) of the index’s father had mild thrombocytopenia and was diagnosed with BMF and 3 acquired TP53 mutations along with this study. Family 3: patient 1438 had spontaneously recovered from aplastic anemia in her childhood. At age 31 years, while pregnant, she was identified as having persistent thrombocytopenia. A next-generation sequencing myeloid gene panel on her peripheral blood sample detected a somatically mutated TP53 clone. BM samples showed severe BMF. (B) Analysis of the validation set. One (patient 1443) of 7 AML M6 patients was found homozygous for ERCC6L2 c.1457delT. Family 4: patient 1443 was age 65 years when diagnosed with AML M6. His sister had died as a result of severe aplastic anemia (or BMF) at a young age. (C) No ERCC6L2 c.1457delT homozygotes were found in the control set of 165 AML patients with other subtypes. Mut, mutated; tAML, therapy-related AML; wt, wild type.
Families in this study and the selection process of patient data. Individuals who have been genotyped for the ERCC6L2 c.1457delT mutation are marked with an asterisk (*). Black dots (•) represent heterozygous carriers of the mutation. Probands in each family are marked with arrows. (A) Families (1-3) in the discovery set. Family 1: patient 1445 was age 38 years when referred to hematologist because of pancytopenia. His BM was dysplastic with strong erythroid predominance and an excess of myeloid blasts. Aiming at allogeneic hematopoietic stem cell transplantation (HSCT), his 36-year-old sister (patient 1458) was examined as a donor candidate. Tests revealed peripheral blood cytopenias. The following BM examination revealed MDS, which quickly progressed to AML M6. She died as a result of refractory leukemia. Patient 1445 underwent HSCT from a registry donor but relapsed quickly with a therapy-resistant AML M6 and died as a result of the disease. Family 2: the index patient (1450) age 18 years was diagnosed with BMF of unknown origin and referred to the hematology department in 2018. Her 2 paternal aunts (patients 1459 and 1439) had died as a result of AML M6. The twin sister (patient 1463) of the index’s father had mild thrombocytopenia and was diagnosed with BMF and 3 acquired TP53 mutations along with this study. Family 3: patient 1438 had spontaneously recovered from aplastic anemia in her childhood. At age 31 years, while pregnant, she was identified as having persistent thrombocytopenia. A next-generation sequencing myeloid gene panel on her peripheral blood sample detected a somatically mutated TP53 clone. BM samples showed severe BMF. (B) Analysis of the validation set. One (patient 1443) of 7 AML M6 patients was found homozygous for ERCC6L2 c.1457delT. Family 4: patient 1443 was age 65 years when diagnosed with AML M6. His sister had died as a result of severe aplastic anemia (or BMF) at a young age. (C) No ERCC6L2 c.1457delT homozygotes were found in the control set of 165 AML patients with other subtypes. Mut, mutated; tAML, therapy-related AML; wt, wild type.
Patients with a homozygous ERCC6L2 mutation
| Family | Patient | Disease course | Age at hematological diagnosis, y | Previous conditions | Family history of malignancies | BM karyotype | Somatic mutations in BM | Somatic mutation analysis | ||
|---|---|---|---|---|---|---|---|---|---|---|
| FLT3/ NPM1 | TP53, VAF | Other | ||||||||
| 1 | 1445 | MDS → AML M6 → relapse soon after allogeneic HSCT; died | MDS at 38; AML M6 at 39 | No | Father died as a result of CRC in his 50s; paternal cousin died of pancreatic cancer in 40s; 1458 | Hypodiploid 41-43, −5, −7 −17, −18, −19, −20 | −/− | c.532C>G p.(His178Asp) 35% | None | Whole-exome sequencing |
| 1 | 1458 | MDS → AML M6 → relapsed on chemotherapy; died | MDS at 36; AML M6 at 37 | Melanoma in situ × 2 (surgery) | Father died as a result of CRC in his 50s; paternal cousin died of pancreatic cancer in 40s; 1445 | Hypodiploid 43, −7, −12, 5q− | −/− | c.517G>A p.(Val173Met) | N/A | Capillary sequencing of TP53 |
| 2 | 1450 | BMF; alive | 14 | No | 2 paternal aunts (1439, 1459) died as a result of AML M6 | CN | −/− | None | None | NGS panel |
| 2 | 1439 | AML M6 → relapsed 13 mo after allogeneic HSCT; died | 59 | No | Sister (1459) died as a result of AML M6; other sister (1463) has BMF | t(3;12;?),t(12;?), −7, −5, t(5;?) | −/− | c.577C>T p.(His193Tyr), c.818G>A p.(Arg273His) | N/A | Capillary sequencing of TP53 |
| 2 | 1459* | AML M6 → refractory disease; died | 38 | No | Sister (1439) died as a result of AML M6; other sister (1463) has BMF | Complex (specific data N/A) | N/A | N/A | N/A | N/A |
| 2 | 1463 | Mild neutropenia and thrombocytopenia → BMF; alive | Cytopenias at 47; BMF at 59 | No | 2 sisters (1459 and 1439) died as a result of AML M6 | CN | −/− | c.743G>A p.(Arg248Gln) 5%, c.830G>T p.(Cys277Phe) 23%, c.843C>A p.(Asp281Glu) 11% | None | NGS panel |
| 3 | 1438 | AA, spontaneous recovery → marginal neutropenia and thrombocytopenia and severe BMF 20 y later | AA at 11; mild cytopenias at 20; BMF at 31 | Cerebral vein thrombosis, Rathke’s cyst | 2 cousins (mother’s side) with some hematological symptoms; grandmother died as a result of leukemia NOS; 6/7 of grandmother’s siblings died of solid tumors; 2/13 of father’s siblings died of cancer (CRC and liver) | CN | −/− | c.659A>G p.(Tyr220Cys) 31% | None | NGS panel |
| 4 | 1443 | AML M6 → relapsed soon after allogeneic HSCT; died | AML M6 at 65 | Tubular adenoma with dysplasia in rectum at 59 | Sister died as a result of AA at 34 | 42-46, del(5)(q31), dup(5)(q31)/t(5;5), −7, 11q23/MLL amplification or translocation, −4 | −/− | c.818G>A p.(Arg273His), c.856G>A p.(Glu286Lys) | N/A | Capillary sequencing of TP53 |
| Family | Patient | Disease course | Age at hematological diagnosis, y | Previous conditions | Family history of malignancies | BM karyotype | Somatic mutations in BM | Somatic mutation analysis | ||
|---|---|---|---|---|---|---|---|---|---|---|
| FLT3/ NPM1 | TP53, VAF | Other | ||||||||
| 1 | 1445 | MDS → AML M6 → relapse soon after allogeneic HSCT; died | MDS at 38; AML M6 at 39 | No | Father died as a result of CRC in his 50s; paternal cousin died of pancreatic cancer in 40s; 1458 | Hypodiploid 41-43, −5, −7 −17, −18, −19, −20 | −/− | c.532C>G p.(His178Asp) 35% | None | Whole-exome sequencing |
| 1 | 1458 | MDS → AML M6 → relapsed on chemotherapy; died | MDS at 36; AML M6 at 37 | Melanoma in situ × 2 (surgery) | Father died as a result of CRC in his 50s; paternal cousin died of pancreatic cancer in 40s; 1445 | Hypodiploid 43, −7, −12, 5q− | −/− | c.517G>A p.(Val173Met) | N/A | Capillary sequencing of TP53 |
| 2 | 1450 | BMF; alive | 14 | No | 2 paternal aunts (1439, 1459) died as a result of AML M6 | CN | −/− | None | None | NGS panel |
| 2 | 1439 | AML M6 → relapsed 13 mo after allogeneic HSCT; died | 59 | No | Sister (1459) died as a result of AML M6; other sister (1463) has BMF | t(3;12;?),t(12;?), −7, −5, t(5;?) | −/− | c.577C>T p.(His193Tyr), c.818G>A p.(Arg273His) | N/A | Capillary sequencing of TP53 |
| 2 | 1459* | AML M6 → refractory disease; died | 38 | No | Sister (1439) died as a result of AML M6; other sister (1463) has BMF | Complex (specific data N/A) | N/A | N/A | N/A | N/A |
| 2 | 1463 | Mild neutropenia and thrombocytopenia → BMF; alive | Cytopenias at 47; BMF at 59 | No | 2 sisters (1459 and 1439) died as a result of AML M6 | CN | −/− | c.743G>A p.(Arg248Gln) 5%, c.830G>T p.(Cys277Phe) 23%, c.843C>A p.(Asp281Glu) 11% | None | NGS panel |
| 3 | 1438 | AA, spontaneous recovery → marginal neutropenia and thrombocytopenia and severe BMF 20 y later | AA at 11; mild cytopenias at 20; BMF at 31 | Cerebral vein thrombosis, Rathke’s cyst | 2 cousins (mother’s side) with some hematological symptoms; grandmother died as a result of leukemia NOS; 6/7 of grandmother’s siblings died of solid tumors; 2/13 of father’s siblings died of cancer (CRC and liver) | CN | −/− | c.659A>G p.(Tyr220Cys) 31% | None | NGS panel |
| 4 | 1443 | AML M6 → relapsed soon after allogeneic HSCT; died | AML M6 at 65 | Tubular adenoma with dysplasia in rectum at 59 | Sister died as a result of AA at 34 | 42-46, del(5)(q31), dup(5)(q31)/t(5;5), −7, 11q23/MLL amplification or translocation, −4 | −/− | c.818G>A p.(Arg273His), c.856G>A p.(Glu286Lys) | N/A | Capillary sequencing of TP53 |
All TP53 mutations reported in NM_000546.5. Variant allele frequency (VAF) not available for capillary sequencing data. Only hotspot exons 5-9 were checked with capillary sequencing (supplemental methods).
AA, aplastic anemia; CN, normal chromosomes; CRC, colorectal cancer; NGS, next-generation sequencing; NOS, not otherwise specified; N/A, not available.
Not tested.
In the series of 7 other AML M6 patients, we found 1 with the same homozygous ERCC6L2 mutation. Clinical characteristics of all ERCC6L2-mutated patients are listed in Table 1 and in the supplemental Data. ERCC6L2 c.1457delT was identified as heterozygous in 3 patients (consistent with gnomAD MAF of .005) in the control set of 165 AMLs of other subtypes. No other ERCC6L2 mutations were present in the AML germ line exomes, nor did we detect any biallelic ERCC6L2 mutations. In summary, 4 of the 10 tested AML M6 cases carried the homozygous ERCC6L2 mutation in comparison with 0 of 165 in the control group of other subtypes of AML (P = 9.734 × 10−5; only statistically independent cases [n = 3] were included). We also investigated germ line ERCC6L2 variants in 10 389 cancer patients (including 142 AML cases) available in The Cancer Genome Atlas PanCanAtlas data set.12 No homozygous protein-truncating rare (<5% minor allele frequency [MAF]) variants were found.
Somatic tumor protein p53 (TP53) mutations are prevalent in AML M6, at 36% compared with 11% in other AML subtypes.9,13 Two of 3 BMF cases and all AML patients with biallelic ERCC6L2 mutation already had acquired somatic TP53 mutations in their BM before AML diagnosis (Table 1). No other somatic mutations in myeloid genes with recurrent mutations in AML or MDS were found.
Median age at diagnosis of AML M6 in our ERCC6L2-mutated patients was 49 years (39 years if including patient 1459). In the other AML M6 patients, median age was 67 years, consistent with the previously reported median of 68 years.13 Despite the lower age at leukemia onset in ERCC6L2-mutated patients, none survived, which reflects the dismal prognosis of AML M6.7 None of the AML M6 patients were known to have BMF preceding leukemia, and no blood count data were available from the time before AML diagnosis; however, relatives of patients 1439 and 1459 reported that both individuals had anemia in their youth. This may be explained by the (symptomless) mild cytopenias sometimes observed in BMF.
Although a notably high penetrance was observed here, a larger sample series is needed for a more refined assessment of penetrance. The identified ERCC6L2 mutation seems to be a founder mutation in Finland (gnomAD Finns MAF = .005 vs global MAF = .0005) and may be enriched in certain areas, explaining the inheritance pattern in family 2 with ancestors from the same region (Figure 1).14
We also identified individuals in earlier phases of the disease continuum from BMF to leukemia. Interestingly, 2 of the 3 BMF patients had somatic TP53 mutations representing clonal evolution. This was also reflected in the ERCC6L2-mutated AML M6 cases, because they were all carriers of 1 or 2 somatic TP53 mutations at the time of leukemia diagnosis, suggesting a strong positive selection. TP53 alterations in AML M6 are not rare, but we suggest that, at least in the setting of ERCC6L2-driven leukemogenesis, they represent the early steps toward malignancy and lead to poor leukemia therapy results. This may be similar to Shwachman-Diamond syndrome, which is another well-known BMF syndrome with strong leukemia predisposition.15,16 The order of molecular changes is in contrast to that of 5q− MDS, for example, where TP53 alterations are thought to occur after chromosomal rearrangements.8 How ERCC6L2 deficiency predisposes individuals to TP53 mutations warrants further study. Notably, recent reports have demonstrated the high impact of somatically mutated TP53 clones in the dynamics of leukemogenesis.17,18
We report a direct association of a homozygous truncating germ line mutation in ERCC6L2 with a specific high-risk leukemia subtype characterized by TP53 mutation(s) and erythroid predominance resembling AML M6 by FAB classification. Certain genes have been previously associated with AML M6 predisposition, but the study families have also presented with other types of leukemias and/or hematological malignancies, indicating a less lineage-restricted predisposition.19,20 To our knowledge, this is the first time a germ line alteration has been suggested to cause a strictly specific subset of acute leukemia. In the era of precision medicine, our findings suggest that AML with somatic TP53 mutations and erythroid predominance stemming from biallelic ERCC6L2 mutations forms a new entity of AML within myeloid neoplasms with germ line predisposition.
Our families have acknowledged for years that AML with a dismal prognosis runs among them. This study has finally discovered the culprit and has also supplied some family members with a relieving verdict. On the basis of our findings and previous reports on ERCC6L2-driven BMF, we suggest hematologists consider careful follow-up and prompt planning of HSCT, which is so far the only potentially lifesaving possibility for ERCC6L2-deficient patients with BMF at risk of leukemia.
The study was approved by the ethics committee of Helsinki University Hospital (#206/13/03/03/2016 and #303/13/03/01/2011). All living participants provided informed written consent to take part in the study.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Lotta Katainen for skillful technical help and text editing, and L. A. Aaltonen and P. Vahteristo for valuable comments on the manuscript. Some of the samples and data used in this project were provided by the Finnish Hematology Registry and Clinical Biobank (FHRB) and Biobank Borealis. The authors thank all the patients for their generous participation.
This study was supported by grants from the Academy of Finland (#274474, #312492, and #284538), Helsinki University Hospital Comprehensive Cancer Center research funding, the Cancer Society of Finland, the Signe and Ane Gyllenberg Foundation, and the Väre Foundation for Pediatric Cancer Research. The FHRB Biobank is supported by the Finnish Association of Hematology, the Finnish Red Cross Blood Service, Institute for Molecular Medicine Finland, and the participating hospitals in Finland.
S.P.M.D. is a PhD candidate at the University of Helsinki; this work is submitted in partial fulfillment of the requirement for the PhD program.
The sponsors of this study are public or nonprofit organizations that support science in general; they had no role in gathering, analyzing, or interpreting the data.
Authorship
Contribution: S.P.M.D. analyzed the whole-exome sequencing data; P.S. and S.P.M.D. performed the capillary sequencing and analyzed the results; P.E.K. reexamined the bone marrow aspirate slides and biopsy specimens; U.W.-K. collected the patient samples and clinical data, together with M.P., S. Kakko, E.-R.S., U.S., and K.O.; S. Kytölä analyzed and interpreted the next-generation sequencing panel data; E.P. performed the statistical analysis and analyzed the online data sets for truncating ERCC6L2 mutations; K.P. provided the control exome data; S.P.M.D. drafted the manuscript; O.K. and U.W.-K. designed the study, supervised the experiments, and finalized the manuscript; and all authors revised and approved the final version of the manuscript.
Conflict-of-interest disclosure: M.P. has had travel, accommodations, and expenses provided by Amgen and Pfizer; U.S. has provided consulting for Mylan; K.O. has had travel, accommodations, and expenses provided by Novartis, Novartis Oncology, and AstraZeneca; and U.W.-K. has provided consulting for Pfizer and Sanofi-Genzyme. The remaining authors declare no competing financial interests.
Correspondence: Outi Kilpivaara, Applied Tumor Genomics Research Program, Faculty of Medicine, University of Helsinki, Biomedicum Helsinki 1, Rm B322b, PO Box 63, 00014 University of Helsinki, Helsinki, Finland; e-mail: outi.kilpivaara@helsinki.fi; and Ulla Wartiovaara-Kautto, Helsinki University Hospital, Comprehensive Cancer Center, Department of Hematology, PO Box 372, 00029 HUS, Helsinki, Finland; e-mail: ulla.wartiovaara-kautto@hus.fi.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal