Key Points
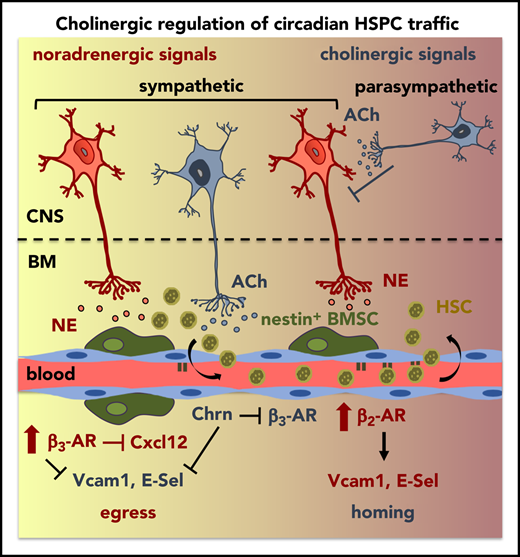
Central parasympathetic signals repress sympathetic tone at night to orchestrate day/night HSPC traffic.
Local sympathetic cholinergic signals modulate HSPC traffic by inhibiting diurnal BM vascular adhesion and β3-adrenergic signaling at night.

Abstract
Hematopoietic stem and progenitor cells (HSPCs) and leukocytes circulate between the bone marrow (BM) and peripheral blood following circadian oscillations. Autonomic sympathetic noradrenergic signals have been shown to regulate HSPC and leukocyte trafficking, but the role of the cholinergic branch has remained unexplored. We have investigated the role of the cholinergic nervous system in the regulation of day/night traffic of HSPCs and leukocytes in mice. We show here that the autonomic cholinergic nervous system (including parasympathetic and sympathetic) dually regulates daily migration of HSPCs and leukocytes. At night, central parasympathetic cholinergic signals dampen sympathetic noradrenergic tone and decrease BM egress of HSPCs and leukocytes. However, during the daytime, derepressed sympathetic noradrenergic activity causes predominant BM egress of HSPCs and leukocytes via β3–adrenergic receptor. This egress is locally supported by light-triggered sympathetic cholinergic activity, which inhibits BM vascular cell adhesion and homing. In summary, central (parasympathetic) and local (sympathetic) cholinergic signals regulate day/night oscillations of circulating HSPCs and leukocytes. This study shows how both branches of the autonomic nervous system cooperate to orchestrate daily traffic of HSPCs and leukocytes.
Introduction
The bone marrow (BM) microenvironment for hematopoietic stem cells (HSCs) includes many cell types that dynamically regulate HSC quiescence, maintenance, activation, proliferation, differentiation, and migration.1 We hypothesized that a master regulator of tissue homeostasis, the autonomic nervous system, might orchestrate different HSC responses to meet physiological demands. This system consists of the enteric nervous system, the sympathetic nervous system (SNS), and the parasympathetic nervous system (PNS). Norepinephrine and acetylcholine are generally postganglionic neurotransmitters of the SNS (noradrenergic) and the PNS (cholinergic). However, some sympathetic neurons switch from the noradrenergic to the cholinergic phenotype during postnatal development in periosteum and salivary glands,2-4 but the role of sympathetic cholinergic fibers in bone has remained elusive.
Sympathetic noradrenergic fibers innervate BM5 and regulate physiological traffic of HSCs and leukocytes, which follow day/night oscillations in mice6,7 and humans.8,9 BM noradrenergic fibers10 and central cholinergic muscarinic signals11 regulate hematopoietic stem and progenitor cell (HSPC) mobilization enforced by granulocyte colony-stimulating factor (G-CSF). However, whether the cholinergic (sympathetic/parasympathetic) nervous system regulates physiological HSPC traffic is unknown. Elucidating this regulation might explain rhythmic HSC and leukocyte traffic and, more importantly, suggest new approaches to therapeutically improve HSC homing/egress.
In mice, the SNS has been reported to be involved in HSC and leukocyte release from BM into circulation during the daytime6 and their BM homing during the evening/night.12 Therefore, it has remained unknown how similar noradrenergic signals can stimulate 2 opposite processes (BM egress and homing) at different times. Therefore, we studied whether the cholinergic nervous system interacts with the noradrenergic nervous system to orchestrate day/night migration of HSCs and leukocytes.
Mice show 2 daily peaks of adrenergic receptor (AR)-mediated HSPC activity: 1 during darkness (when mice are more active) and another following light exposure (when the resting period starts).13 This is consistent with the observed day/night fluctuations of their ligands norepinephrine (noradrenergic) and epinephrine (adrenergic) in murine/human plasma.14,15 Whereas norepinephrine is the principal sympathetic neurotransmitter released in BM, blood epinephrine derives mainly from the adrenal glands. Furthermore, epinephrine and norepinephrine exhibit opposite >30-fold higher affinities for β2-AR and β3-AR, respectively.16 Thus, we hypothesized that norepinephrine and epinephrine might play differential roles in HSPC and leukocyte migration by activating different β-ARs.
To understand this complex regulatory pathway, we have analyzed HSPC and leukocyte traffic in rodents with impaired cholinergic neurotransmission during day/night cycles. We have uncovered a dual cholinergic regulation of the rhythmic migration of HSPCs and leukocytes. At night, central parasympathetic signals antagonize sympathetic noradrenergic activity to restrict HSPC egress. Parasympathetic cholinergic regulation cooperates with endocrine epinephrine–β2-AR signaling to trigger BM homing. Light exposure acutely induces sympathetic activity and suppresses parasympathetic tone.17,18 Consequently, dawn triggers sympathetic release of norepinephrine (noradrenergic) and acetylcholine (cholinergic) in the BM, which activate β3-AR signaling and inhibit vascular adhesion, respectively. This concerted action in the morning reduces BM homing and permits daily egress of HSPCs and leukocytes. Therefore, this study shows how central (parasympathetic) and peripheral (sympathetic) cholinergic neural signals regulate physiological migration of HSCs and leukocytes.
Methods
Age- and sex-matched Gfra2−/−,19 Nes-gfp20 (gift from G.E. Enikolopov), Adrb2tm1Bkk/J21 (gift from G. Karsenty), Adrb3tm1Lowl, B6;129X1-Nrtntm1Jmi/J (Nrtn−/−),22 and congenic CD45.1 and CD45.2 C57BL/6J mice (The Jackson Laboratory) were used in this study. Mice were housed in specific pathogen–free facilities. All experiments followed protocols approved by the Animal Welfare Ethical Committees at Centro Nacional de Investigaciones Cardiovaculares (CNIC) and the University of Cambridge (PPL 70/8406) and were compliant with European Union recommendations. Detailed procedures for in vivo experiments, flow cytometry, cell culture, long-term competitive repopulation assays, homing assays, immunofluorescence, enzyme-linked immunosorbent assays, and quantitative real-time polymerase chain reaction are available in supplemental Methods, available on the Blood Web site.
Results
The PNS inhibits sympathetic noradrenergic activity centrally to reduce circulating HSPCs at night
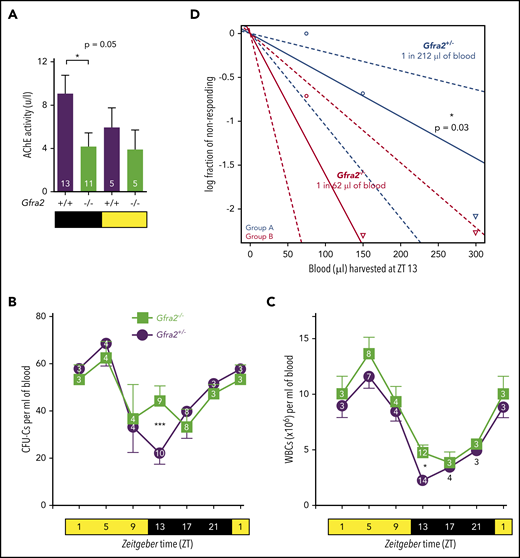
The GDNF family receptor α 2 (GFRα2) promotes the development and survival of cholinergic neurons (parasympathetic or sympathetic).19,23 Therefore, we used Gfra2−/− mice as a model to study the cholinergic regulation of HSPC traffic. Gfra2−/− mice have deficient parasympathetic cholinergic innervation of lacrimal and salivary glands, small bowel,19 endocrine pancreas,24 and reproductive organs.25 Interestingly, parasympathetic activity follows daily oscillations in mice, with increased activity during the day–night shift in stomach,26 lungs,27 and heart.28,29 Consistent with these studies, we found that acetylcholinesterase (AChE) cholinergic activity appeared higher in wild-type (WT) mice urine collected from nightshift to morning compared with daytime harvest (Figure 1A). Urine AChE activity was halved in Gfra2−/− mice at night, suggesting widespread deficient parasympathetic activity in Gfra2−/− mice manifesting at night.
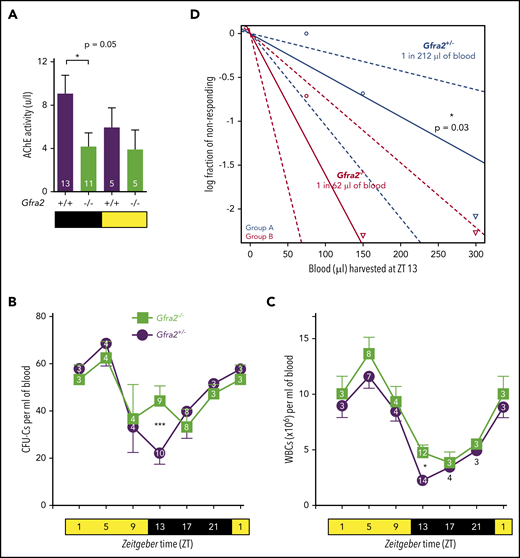
Cholinergic neural signals regulate circadian traffic of HSCs and leukocytes in mice. (A) AChE activity in urine samples from Gfra2−/− and WT mice collected in the nocturnal (black) and diurnal (yellow) periods. HSPCs measured as CFU-Cs (B) and WBCs (C) in peripheral blood of Gfra2−/− mice and control Gfra2+/− mice harvested at the specified ZT (hours after light onset). ZT1 has been duplicated to facilitate viewing. (D) HSCs, measured by long-term competitive repopulation assay, in peripheral blood harvested at ZT13 from Gfra2−/− mice and control Gfra2+/− mice. The log fraction of mice that failed reconstitution is plotted against the transplanted blood volume using ELDA software.59 Likelihood ratio test of single-hit model, P = .006, χ2 test. Blood HSC concentrations are indicated (n = 5). Data are mean ± standard error of the mean; n (inside bars/symbols) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, ***P < .001, 1-way ANOVA and Bonferroni comparisons (A), multiple 2-tailed test (B-C), χ2 test (D).
Cholinergic neural signals regulate circadian traffic of HSCs and leukocytes in mice. (A) AChE activity in urine samples from Gfra2−/− and WT mice collected in the nocturnal (black) and diurnal (yellow) periods. HSPCs measured as CFU-Cs (B) and WBCs (C) in peripheral blood of Gfra2−/− mice and control Gfra2+/− mice harvested at the specified ZT (hours after light onset). ZT1 has been duplicated to facilitate viewing. (D) HSCs, measured by long-term competitive repopulation assay, in peripheral blood harvested at ZT13 from Gfra2−/− mice and control Gfra2+/− mice. The log fraction of mice that failed reconstitution is plotted against the transplanted blood volume using ELDA software.59 Likelihood ratio test of single-hit model, P = .006, χ2 test. Blood HSC concentrations are indicated (n = 5). Data are mean ± standard error of the mean; n (inside bars/symbols) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, ***P < .001, 1-way ANOVA and Bonferroni comparisons (A), multiple 2-tailed test (B-C), χ2 test (D).
Murine HSPCs and leukocytes are preferentially released into the circulation during the day,6 whereas more white blood cells (WBCs) home to BM at night.12 We quantified WBCs and circulating HSPCs, measured as colony-forming units in culture (CFU-Cs), over 24 hours. Control mice exhibited normal oscillations of circulating HSPCs and WBCs.6 These cells peaked 5 hours after light onset [at Zeitgeber time (ZT)5; morning] and reached similar nadirs early at night (ZT13) in WT mice and Gfra2+/− mice (Figure 1B-C; supplemental Figure 1A), which were used as control littermate mice in most experiments. In contrast, Gfra2−/− mice showed normal daytime values but exhibited two- to threefold more circulating HSPCs and WBCs at ZT13 (Figure 1B-C), whereas other blood parameters remained unchanged (supplemental Figure 1B-E). To measure circulating HSCs, we performed competitive long-term repopulation assays using limiting dilutions of blood harvested at ZT13. Gfra2−/− mice showed 3.5 times more circulating HSCs early at night (Figure 1D). Despite this difference, BM nucleated cells and BM HSPCs remained unchanged (supplemental Figure 1F-G), as expected, because their numbers are much higher in BM than in bloodstream. The frequency of leukocyte subsets (supplemental Figure 2) and other cell types (data not shown) appeared normal in Gfra2−/− blood and BM, excluding differentiation defects. These results show a time-specific accumulation of circulating HSPCs and WBCs in cholinergic neural–deficient mice at night.
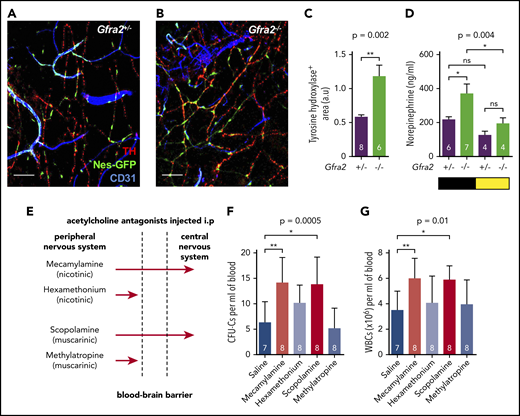
Previous studies have suggested that sympathetic noradrenergic fibers contribute to BM HSC egress,6,7 G-CSF–induced mobilization,10 and WBC BM homing.12 We stained tyrosine hydroxylase–positive noradrenergic fibers and found them doubled in Gfra2−/− BM (Figure 2A-C). In addition, urine concentration of norepinephrine was doubled in Gfra2−/− mice at night (Figure 2D), suggesting that parasympathetic deficiency in Gfra2−/− mice derepresses noradrenergic activity at night, possibly triggering abnormal nocturnal release of HSPCs and leukocytes. To investigate whether cholinergic signals repress noradrenergic activity centrally or peripherally, we injected WT mice intraperitoneally (i.p.) with blood–brain barrier (BBB)-permeable or BBB-nonpermeable cholinergic (acetylcholine) antagonists (Figure 2E). Only BBB-permeable antagonists (mecamylamine and scopolamine) increased circulating HSPCs and WBCs at ZT13 (Figure 2F-G), suggesting a central inhibition of sympathetic tone by the PNS. This result expands the role of the central cholinergic nervous system from enforced HSC mobilization11 to the regulation of physiological traffic of HSPCs and leukocytes. Therefore, central parasympathetic inhibition of sympathetic tone reduces BM release of HSPCs and leukocytes at night.
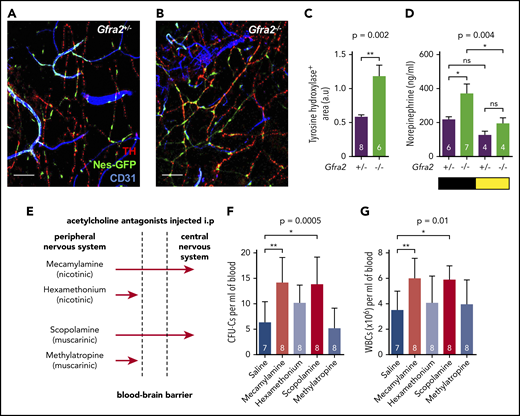
Cholinergic neural signals regulate HSPC and leukocyte traffic by modulating sympathetic noradrenergic tone centrally. (A-B) Representative immunofluorescence of CD31+ endothelial cells (blue), tyrosine hydroxylase–positive sympathetic nerve fibers (TH; red), and nestin-GFP+ cells (green) in the skull BM of Nes-gfp;Gfra2+/− and Nes-gfp;Gfra2−/− compound mice. Scale bars, 100 μm. (C) Quantification of the skull BM area covered by TH+ sympathetic noradrenergic nerve fibers from Gfra2+/− mice and Gfra2−/− mice. (D) Nocturnal (black) and diurnal (yellow) norepinephrine concentration in the urine of Gfra2+/− and Gfra2−/− compound mice. (E) Scheme illustrating the different types of cholinergic antagonists used and their capacity to cross the BBB. Mecamylamine and scopolamine are BBB-permeable antagonists, whereas hexamethonium and methylatropine are BBB-nonpermeable antagonists. Blood-circulating HSPCs, measured as CFU-Cs (F) and WBCs (G) at ZT13 in WT mice treated with acetylcholine antagonists (i.p.) at ZT5. (C-D,F-G) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, unpaired 2-tailed t test (C), 1-way analysis of variance with Bonferroni comparisons (D,F-G). a.u, arbitrary units; ns, not significant.
Cholinergic neural signals regulate HSPC and leukocyte traffic by modulating sympathetic noradrenergic tone centrally. (A-B) Representative immunofluorescence of CD31+ endothelial cells (blue), tyrosine hydroxylase–positive sympathetic nerve fibers (TH; red), and nestin-GFP+ cells (green) in the skull BM of Nes-gfp;Gfra2+/− and Nes-gfp;Gfra2−/− compound mice. Scale bars, 100 μm. (C) Quantification of the skull BM area covered by TH+ sympathetic noradrenergic nerve fibers from Gfra2+/− mice and Gfra2−/− mice. (D) Nocturnal (black) and diurnal (yellow) norepinephrine concentration in the urine of Gfra2+/− and Gfra2−/− compound mice. (E) Scheme illustrating the different types of cholinergic antagonists used and their capacity to cross the BBB. Mecamylamine and scopolamine are BBB-permeable antagonists, whereas hexamethonium and methylatropine are BBB-nonpermeable antagonists. Blood-circulating HSPCs, measured as CFU-Cs (F) and WBCs (G) at ZT13 in WT mice treated with acetylcholine antagonists (i.p.) at ZT5. (C-D,F-G) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, unpaired 2-tailed t test (C), 1-way analysis of variance with Bonferroni comparisons (D,F-G). a.u, arbitrary units; ns, not significant.
Cholinergic signaling regulates HSPC BM homing by modulating β2-AR–dependent BM vascular adhesion
Next, we investigated whether alterations in nocturnal BM homing in Gfra2−/− mice could further explain the accumulation of circulating HSPCs and WBCs. We transplanted donor cells at ZT10 (evening) using Gfra2−/− mice and control Gfra2+/− mice as donors/recipients. We analyzed recipient mice at ZT2 (early morning) to measure overall nocturnal BM homing (Figure 3A), which appeared normal in myeloablated mice (Figure 3B). However, because lethal irradiation enforces BM homing and might mask differences, we independently transplanted nonirradiated mice (Figure 3C). Surprisingly, nocturnal HSPC BM homing was not impaired (as it would be expected from HSPCs accumulating in circulation) but instead doubled in nonmyeloablated Gfra2−/− mice (Figure 3D; supplemental Figure 3). Homing of donor Gfra2−/− HSPCs in control recipient mice was normal (Figure 3D), indicating noncell-autonomous cholinergic regulation of HSPC homing, which we investigated next at the molecular level.

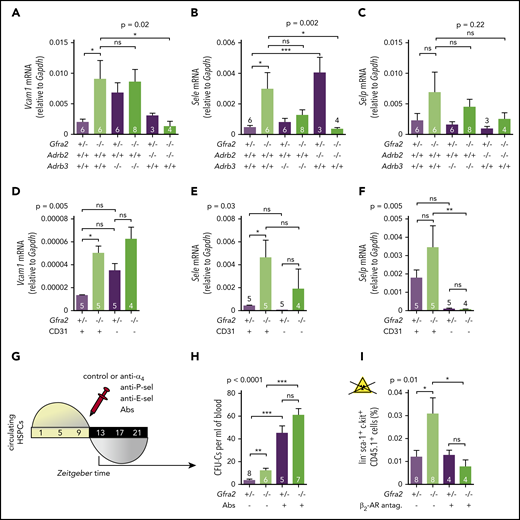
The PNS regulates nocturnal HSC BM adhesion and homing. (A) Scheme showing the protocol used for the HSPC BM homing assay with irradiation (12 Gy). (B) Frequencies of HSPCs (homing efficiency) at ZT2 that homed during the night to the BM after IV transplantation into lethally irradiated mice (12 Gy) (to promote homing) at ZT10. Gfra2−/− mice and control Gfra2+/− mice were used as donor (lower genotypes) or recipients (upper genotypes) in all combinations. Homing efficiency is determined as the percentage of CFU-Cs obtained from BM harvested from irradiated mice in comparison with CFU-Cs obtained from a nonirradiated mouse. (C) Scheme showing the protocol used for the HSPC BM homing assay without irradiation. (D) Frequencies of donor-derived Gfra2+/+ or Gfra2−/− lin−sca-1+c-kit+ HSPCs (identified by flow cytometry) at ZT2 that homed during the night to the BM after IV transplantation in nonirradiated congenic mice at ZT10. Vcam1 (E), Sele (F), and Selp (G) mRNA expression in the unfractionated BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (B,D-G) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, 1-way analysis of variance and Bonferroni comparisons (B,D), multiple 2-tailed test (E-G). ns, not significant.
The PNS regulates nocturnal HSC BM adhesion and homing. (A) Scheme showing the protocol used for the HSPC BM homing assay with irradiation (12 Gy). (B) Frequencies of HSPCs (homing efficiency) at ZT2 that homed during the night to the BM after IV transplantation into lethally irradiated mice (12 Gy) (to promote homing) at ZT10. Gfra2−/− mice and control Gfra2+/− mice were used as donor (lower genotypes) or recipients (upper genotypes) in all combinations. Homing efficiency is determined as the percentage of CFU-Cs obtained from BM harvested from irradiated mice in comparison with CFU-Cs obtained from a nonirradiated mouse. (C) Scheme showing the protocol used for the HSPC BM homing assay without irradiation. (D) Frequencies of donor-derived Gfra2+/+ or Gfra2−/− lin−sca-1+c-kit+ HSPCs (identified by flow cytometry) at ZT2 that homed during the night to the BM after IV transplantation in nonirradiated congenic mice at ZT10. Vcam1 (E), Sele (F), and Selp (G) mRNA expression in the unfractionated BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (B,D-G) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, 1-way analysis of variance and Bonferroni comparisons (B,D), multiple 2-tailed test (E-G). ns, not significant.
Firm adhesion to endothelium is the first step during BM homing30 and requires endothelial selectins and vascular cell adhesion protein 1 (Vcam1).31-33 Moreover, expression of Vcam1 and Sele (E-selectin) messenger RNA (mRNA) peaks at night, when HSPCs and WBCs preferentially home to BM.12 In control mice, we confirmed day/night oscillations of Vcam1 and Sele mRNA peaking at ZT13. P-selectin (Selp) showed a similar trend (Figure 3E-G). Interestingly, Gfra2−/− BM showed higher mRNA expression of Vcam1 (4.5-fold) and Sele (6.2-fold) at night (Figures 3E-F, 4A-B). Selp showed a similar, but nonsignificant, trend (Figures 3G, 4C). Deregulated adhesion molecule expression specifically affected endothelial cells, which also exhibited enriched Sele and Selp mRNA expression compared with nonendothelial stromal cells (Figure 4D-F). Flow cytometry confirmed increased Vcam1 protein in endothelial cells from Gfra2−/− mice at ZT13 (supplemental Figure 4A). To test whether increased adhesion explains enhanced BM homing in Gfra2−/− mice, we injected Gfra2−/− mice and control Gfra2+/− mice with control IgG or blocking antibodies against integrin α4 (part of VLA-4, receptor for Vcam1), E-selectin, and P-selectin before the peak of homing (ZT11; Figure 4G). Adhesion-blocking antibodies increased circulating HSPCs in Gfra2−/− mice and control Gfra2+/− mice (as expected) but cancelled out the nocturnal differences between them (Figure 4H). Therefore, enhanced nocturnal HSPC homing in Gfra2−/− mice is caused by increased BM vascular adhesion.
Parasympathetic deficiency increases nocturnal HSPC BM adhesion and homing through β2-adrenergic signaling in the microenvironment.Vcam1 (A), Sele (B), and Selp (C) mRNA expression at ZT13 in the unfractionated BM of control Gfra2+/− mice, Gfra2−/− mice, single β2-AR (Adrb2)-deficient or β3-AR (Adrb3)-deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. Vcam1 (D), Sele (E), and Selp (F) mRNA expression at ZT13 in CD45−Ter119− endothelial (CD31+) or nonendothelial (CD31−) cells from Gfra2−/− and control Gfra2+/− mice. (G) Scheme showing the protocol used for blockade of in vivo HSPC adhesion to blood vessels using antibodies against α4-integrin, P-selectin, and E-selectin (IV injection at ZT11 and analysis at ZT13). (H) HSPCs circulating at ZT13, 2 hours after injection of blocking antibodies (Abs) or control IgG. Please note that CFU-C fold change goes from a 3.2-fold increase in control immunoglobulin G–treated mice to a 1.3-fold increase in blocking antibody–treated mice. (I) Frequencies of donor-derived WT CD45.1+ lin−sca1+ckit+ HSPCs at ZT2 that homed during the night to the BM after IV transplantation (at ZT10) into nonirradiated Gfra2−/− mice or control Gfra2+/− mice preconditioned with saline or β2 adrenergic antagonist (ICI118,551) 4 hours before transplantation. (A-F,H-I) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, 1-way analysis of variance with Bonferroni comparisons. ns, not significant.
Parasympathetic deficiency increases nocturnal HSPC BM adhesion and homing through β2-adrenergic signaling in the microenvironment.Vcam1 (A), Sele (B), and Selp (C) mRNA expression at ZT13 in the unfractionated BM of control Gfra2+/− mice, Gfra2−/− mice, single β2-AR (Adrb2)-deficient or β3-AR (Adrb3)-deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. Vcam1 (D), Sele (E), and Selp (F) mRNA expression at ZT13 in CD45−Ter119− endothelial (CD31+) or nonendothelial (CD31−) cells from Gfra2−/− and control Gfra2+/− mice. (G) Scheme showing the protocol used for blockade of in vivo HSPC adhesion to blood vessels using antibodies against α4-integrin, P-selectin, and E-selectin (IV injection at ZT11 and analysis at ZT13). (H) HSPCs circulating at ZT13, 2 hours after injection of blocking antibodies (Abs) or control IgG. Please note that CFU-C fold change goes from a 3.2-fold increase in control immunoglobulin G–treated mice to a 1.3-fold increase in blocking antibody–treated mice. (I) Frequencies of donor-derived WT CD45.1+ lin−sca1+ckit+ HSPCs at ZT2 that homed during the night to the BM after IV transplantation (at ZT10) into nonirradiated Gfra2−/− mice or control Gfra2+/− mice preconditioned with saline or β2 adrenergic antagonist (ICI118,551) 4 hours before transplantation. (A-F,H-I) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, 1-way analysis of variance with Bonferroni comparisons. ns, not significant.
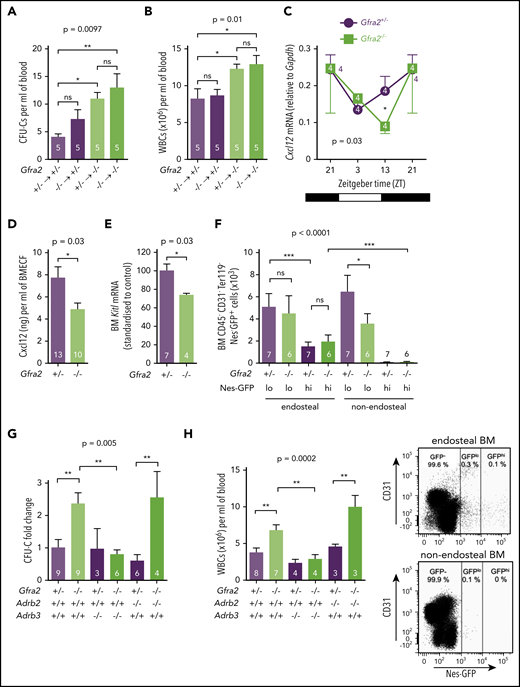
Nocturnal induction of these adhesion molecules by noradrenergic signals promotes WBC BM homing mainly through β2-AR.12 To dissect β-AR function in enhanced BM homing in cholinergic neural–deficient mice, we crossed Gfra2−/− mice with mice lacking the β-ARs involved in HSPC/leukocyte traffic (β2-AR and β3-AR).34 The five- to sixfold increased mRNA expression of Vcam1 and Sele found in Gfra2−/− mice normalized in Gfra2−/−;Adrb2−/− mice (Figure 4A-B). To confirm the role of β2-AR, we measured HSPC homing in recipient mice previously injected with β2-AR antagonist. This treatment normalized BM HSPC homing in Gfra2−/− mice (Figure 4I; supplemental Figure 4B), suggesting that increased sympathetic noradrenergic activity in cholinergic neural–deficient mice promotes HSPC BM homing through β2-AR.
Exacerbated sympathetic noradrenergic activity in cholinergic neural–deficient mice causes abnormal BM egress of HSPCs and leukocytes via β3-ARs
Despite increased BM homing, cholinergic neural–deficient mice accumulate circulating HSPCs and WBCs at night (Figure 1B-C). Therefore, we hypothesized that abnormal nocturnal BM egress might override increased BM homing in Gfra2−/− mice. The Cxcl12-Cxcr4 axis regulates HSPC and leukocyte migration.35,36 We have previously shown that light-triggered sympathetic signals decrease BM Cxcl12 expression and permit HSPC egress into the circulation.6 Therefore, we investigated whether exacerbated sympathetic noradrenergic activity in Gfra2−/− mice (in the absence of inhibitory cholinergic signals) causes abnormal nocturnal BM release of HSPCs and leukocytes. To dissect hematopoietic cell–autonomous and noncell-autonomous effects, we generated chimeric mice through long-term reciprocal BM transplantations. Control recipients of Gfra2−/− hematopoietic cells showed normal WBCs. In contrast, Gfra2−/− recipient mice showed nocturnal accumulation of circulating HSPCs and WBCs (Figure 5A-B), suggesting noncell-autonomous defects in HSPCs and leukocyte traffic in Gfra2−/− mice.
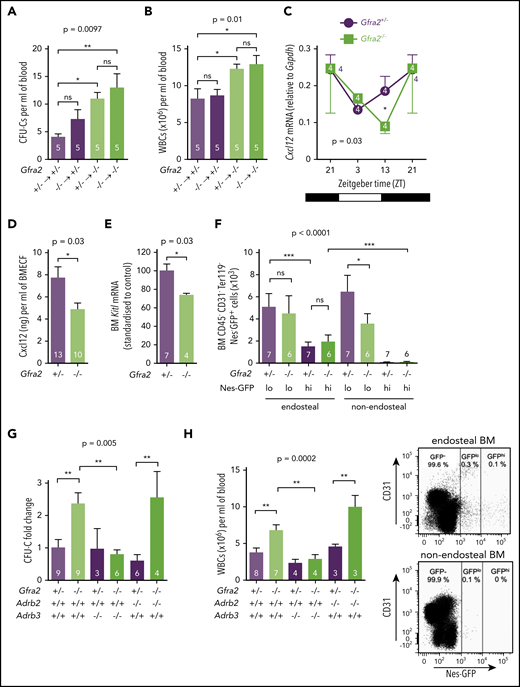
The PNS inhibits β3-adrenergic–dependent BM egress of HSPCs at night. HSPCs, measured as CFU-Cs (A), and WBCs (B) circulating at ZT13, 16 weeks after BM transplantation into lethally irradiated mice. Gfra2−/− mice and control Gfra2+/− mice were used as donor (lower genotypes) or recipients (upper genotypes) in all combinations. (C) Cxcl12 mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (D) Cxcl12 concentration in BM extracellular fluid (BMECF) at ZT13. (E) Kitl mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at ZT13. (F) Number of stromal Nes-GFPhi/lo cells in endosteal and nonendosteal BM (upper panel). Representative flow cytometry plot showing CD31 and Nes-GFP expression in CD45−Ter119− cells isolated from endosteal BM of Gfra2−/− and control Gfra2+/+ mice (lower panel). (G) CFU-C fold change at ZT13 in control Gfra2+/− mice, Gfra2−/− mice, single β2- or β3-AR (Adrb2, Adrb3)–deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. (H) WBCs circulating at ZT13 in control Gfra2+/− mice, Gfra2−/− mice, single β2- or β3-AR (Adrb2, Adrb3)–deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. All data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, 1-way analysis of variance with Bonferroni comparisons (A-B,F-H), multiple 2-tailed test (C), unpaired 2-tailed t test (D-E). ns, not significant.
The PNS inhibits β3-adrenergic–dependent BM egress of HSPCs at night. HSPCs, measured as CFU-Cs (A), and WBCs (B) circulating at ZT13, 16 weeks after BM transplantation into lethally irradiated mice. Gfra2−/− mice and control Gfra2+/− mice were used as donor (lower genotypes) or recipients (upper genotypes) in all combinations. (C) Cxcl12 mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (D) Cxcl12 concentration in BM extracellular fluid (BMECF) at ZT13. (E) Kitl mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at ZT13. (F) Number of stromal Nes-GFPhi/lo cells in endosteal and nonendosteal BM (upper panel). Representative flow cytometry plot showing CD31 and Nes-GFP expression in CD45−Ter119− cells isolated from endosteal BM of Gfra2−/− and control Gfra2+/+ mice (lower panel). (G) CFU-C fold change at ZT13 in control Gfra2+/− mice, Gfra2−/− mice, single β2- or β3-AR (Adrb2, Adrb3)–deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. (H) WBCs circulating at ZT13 in control Gfra2+/− mice, Gfra2−/− mice, single β2- or β3-AR (Adrb2, Adrb3)–deficient mice, or compound Gfra2−/−Adrb2−/− and Gfra2−/−Adrb3−/− mice. All data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, 1-way analysis of variance with Bonferroni comparisons (A-B,F-H), multiple 2-tailed test (C), unpaired 2-tailed t test (D-E). ns, not significant.
Light-triggered sympathetic activity reduces BM Cxcl12 expression and permits HSPC egress.6,37 We found that increased circulating HSPCs and WBCs correlated with reduced Cxcl12 mRNA and protein expression in Gfra2−/− BM at the same time, whereas Cxcr4 expression was unchanged (Figure 5C-D; supplemental Figure 5). Stem cell factor (Kitl) also showed reduced mRNA expression in Gfra2−/− BM during this period (Figure 5E). Cxcl12 and Kitl are highly expressed by nestin+ BM mesenchymal stem/progenitor cells (BMSCs), which regulate HSPC migration.38 Therefore, we intercrossed Gfra2−/− mice with Nes-gfp mice to measure nestin+ BMSCs. We dissected different Nes-GFP+ subpopulations39 by mechanical separation of endosteal/nonendosteal cells.40 Nes-GFPhi cells have been found associated with central arterioles and endosteal transition zone vessels connecting arterioles with sinusoids.39,41,42 Nes-GFPlo cells coincide with Lepr+ cells in nonendosteal sinusoids,43 through which HSPCs and leukocytes transmigrate.41 Whereas Nes-GFPhi cells were unchanged, Nes-GFPlo cells were reduced 40% in nonendosteal Gfra2−/− BM (Figure 5F). Therefore, decreased Cxcl12-dependent HSPC retention at ZT13 correlates with reduced nonendosteal Nes-GFPlo cells in Gfra2−/− BM. Whereas this cholinergic regulation clearly involves nestin+ BMSCs and endothelial cells, it might also target other BM cells.
Because β-adrenergic signaling regulates Cxcl12 expression in nestin+ cells,6 we studied its possible role in enforced BM egress in cholinergic neural–deficient mice. For that purpose, we measured circulating HSPCs and WBCs in Gfra2−/− mice and compound Gfra2−/−;Adrb2−/− or Gfra2−/−;Adrb3−/− mice. Importantly, nocturnal (ZT13) circulating HSPCs and WBCs increased similarly in Gfra2−/− mice and Gfra2−/−;Adrb2−/− mice but normalized in Gfra2−/−;Adrb3−/− mice (Figure 5G-H). Thus, in contrast with β2-AR–regulated BM homing, abnormal nocturnal β3-AR activation reduces Cxcl12 expression and triggers BM egress of HSPCs and leukocytes in cholinergic neural–deficient mice.
BM expression of β2-AR and β3-AR follows day/night oscillations, which are altered in cholinergic neural–deficient mice
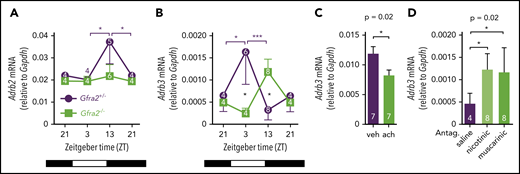
Because β3-AR and β2-AR respectively mediated the egress and homing of BM HSPCs and leukocytes, we hypothesized that expression of β-ARs might follow day/night oscillations regulated by cholinergic signals. We measured BM mRNA expression of β2-AR (Adrb2) and β3-AR (Adrb3) at different time points during the day/night and found oscillations in control mice. Whereas Adrb2 mRNA expression was higher at night, when BM homing becomes predominant, Adrb3 mRNA expression was increased during the day, when β3-AR triggers HSC egress (Figure 6A-B). In contrast, day/night oscillations of Adrb2 were blunted and those of Adrb3 mRNA were inverted in Gfra2−/− BM. Notably, BM Adrb3 mRNA expression was increased threefold in Gfra2−/− mice at night (Figure 6B), further indicating that exacerbated β3-adrenergic signaling at night enforces BM release of HSPCs and leukocytes, possibly explaining why BM egress overrides BM homing in cholinergic neural–deficient mice.
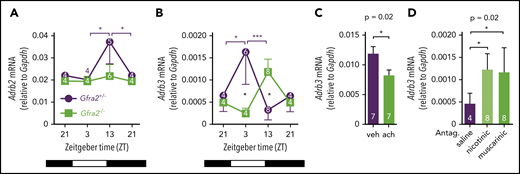
Local cholinergic signals regulate oscillatory expression of β3-AR in BM.Adrb2 (A) and Adrb3 (B) mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (C) Adrb3 mRNA expression in MS-5 cell line cultures treated with vehicle (veh) or acetylcholine (ach; 10 µM) for 6 hours. (D) Adrb3 mRNA expression in the BM of WT mice treated with acetylcholine antagonists (i.p.) at ZT5 and analyzed at ZT13. All data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, ***P < .001, multiple 2-tailed test (A-B), unpaired 2-tailed t test (C), 1-way analysis of variance with Bonferroni comparisons (D).
Local cholinergic signals regulate oscillatory expression of β3-AR in BM.Adrb2 (A) and Adrb3 (B) mRNA expression in the BM of Gfra2−/− and Gfra2+/− control mice at the specified ZT. ZT21 has been duplicated to facilitate viewing. (C) Adrb3 mRNA expression in MS-5 cell line cultures treated with vehicle (veh) or acetylcholine (ach; 10 µM) for 6 hours. (D) Adrb3 mRNA expression in the BM of WT mice treated with acetylcholine antagonists (i.p.) at ZT5 and analyzed at ZT13. All data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, ***P < .001, multiple 2-tailed test (A-B), unpaired 2-tailed t test (C), 1-way analysis of variance with Bonferroni comparisons (D).
Sympathetic cholinergic fibers modulate migratory oscillations by locally regulating the expression of vascular adhesion molecules and β-ARs
Altered BM expression of vascular adhesion molecules (Figures 3E-G, 4A-F) and β-ARs (Figure 6A-B) in cholinergic neural–deficient mice suggested their local regulation by cholinergic signals. Supporting this possibility, treatment with acetylcholine reduced Adrb3 (but not Adrb2) mRNA expression in MS-5 stromal cells (Figure 6C; supplemental Figure 6A). Mirroring the in vitro results, in vivo i.p. administration of acetylcholine antagonists increased BM Adrb3 (but not Adrb2) mRNA expression at night (Figure 6D; supplemental Figure 6B). These results suggest that local cholinergic signals reduce nocturnal β3-AR signaling, which contributes to reduce BM egress of HSPCs and leukocytes. However, only very few studies (focused on bone turnover) have investigated this possible local cholinergic regulation. Previous studies have suggested that cholinergic signals might have bone anabolic effects by antagonizing the SNS but have not agreed between central and local effects.44,45 A recent study showed that central cholinergic signals regulate the hypothalamus-hypophysis axis and G-CSF–induced HSPC migration.11
To study local cholinergic signals, we immunostained skull bones of WT and Gfra2−/− mice for vesicular acetylcholine transporter (VAChT), a validated marker of periosteal sympathetic cholinergic fibers.23 VAChT+ sympathetic cholinergic fibers were present in WT skull but were reduced three- to fourfold in Gfra2−/− skull (Figure 7A-C). Binding of the neurotrophic factor neurturin (Nrtn)22 to its receptor GFRα219,23 promotes development and survival of cholinergic neurons. Therefore, we used Nrtn−/− mice as an additional model to study the cholinergic regulation of HSPC traffic. For verification, we stained WT/Nrtn−/− skull for Gfrα2 (another validated marker of periosteal sympathetic cholinergic fibers).23 In contrast with Gfra2−/− mice, Nrtn−/− mice exhibited doubled periosteal cholinergic fibers (Figure 7D-F) and unchanged BM noradrenergic innervation (supplemental Figure 7A-C) and Cxcl12 content (supplemental Figure 7D). Thus, Nrtn−/− mice exhibit increased sympathetic cholinergic fibers in bone, but apparently normal central sympathetic/parasympathetic activity (supplemental Table 1). Therefore, Gfra2−/− mice and Nrtn−/− mice are loss- and gain-of-function models of bone sympathetic cholinergic innervation, respectively. These opposite phenotypes are likely explained by compensatory threefold upregulation of Gfra2 mRNA in Nrtn−/− BM (Figure 7G) and potential Gfrα2 activation by other neurotrophic factors, as previously reported.46-48 Supporting this compensatory ligand/receptor upregulation, Gfra2−/− mice exhibited doubled BM Nrtn mRNA expression (Figure 7H). Nocturnal WBCs were unchanged in Nrtn−/− mice (Figure 7I), consistent with normal noradrenergic innervation and Cxcl12 expression in these mice.

Sympathetic cholinergic signals locally repress adhesion to BM vessels during the daytime. Z-stack projection showing immunofluorescence of CD31+ endothelial cells (blue) and VAChT+ nerve fibers (red) in the skull of WT (A) and Gfra2−/− (B) mice. Scale bar, 100 μm. (C) Quantification of VAChT+ fibers in the skull periosteum of Gfra2−/− and WT mice. Immunofluorescence of CD31+ endothelial cells (blue) and Gfrα2+ nerve fibers (red) in the skull of WT (D) and Nrtn−/− (E) mice. Scale bar, 100 μm. (F) Quantification of Gfrα2+ fibers in the skull periosteum of Nrtn−/− and WT mice. Gfra2 (G) and Nrtn (H) mRNA expression at ZT13 in the BM of Gfra2−/−, Nrtn−/−, and WT mice. (I) Normalized WBC counts in peripheral blood of Gfra2−/−, Nrtn−/−, and WT mice at ZT13. Vcam1 (J) and Sele (K) mRNA expression at ZT13 in the unfractionated BM of control Nrtn+/+ and compound Nrtn−/− mice. (L) Frequencies of donor-derived WT lin−sca1+ckit+ HSPCs that homed to the BM at ZT5, 6 hours after IV transplantation into nonirradiated Gfra2−/−, Nrtn−/−, and WT mice (at ZT23). (M) Blood CFU-C fold change at ZT5 in WT mice treated with saline, β-AR antagonists, or cholinergic nicotinic (Chrn) antagonist at ZT23. Vcam1 (N) and Sele (O) mRNA expression at ZT5 in the BM of WT mice treated with saline, β-AR antagonists, or cholinergic nicotinic antagonist at ZT23. (C,F-O) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, unpaired 2-tailed t test (C,F,J-K), 1-way analysis of variance with Bonferroni comparisons (G-I,L-O). ns, not significant.
Sympathetic cholinergic signals locally repress adhesion to BM vessels during the daytime. Z-stack projection showing immunofluorescence of CD31+ endothelial cells (blue) and VAChT+ nerve fibers (red) in the skull of WT (A) and Gfra2−/− (B) mice. Scale bar, 100 μm. (C) Quantification of VAChT+ fibers in the skull periosteum of Gfra2−/− and WT mice. Immunofluorescence of CD31+ endothelial cells (blue) and Gfrα2+ nerve fibers (red) in the skull of WT (D) and Nrtn−/− (E) mice. Scale bar, 100 μm. (F) Quantification of Gfrα2+ fibers in the skull periosteum of Nrtn−/− and WT mice. Gfra2 (G) and Nrtn (H) mRNA expression at ZT13 in the BM of Gfra2−/−, Nrtn−/−, and WT mice. (I) Normalized WBC counts in peripheral blood of Gfra2−/−, Nrtn−/−, and WT mice at ZT13. Vcam1 (J) and Sele (K) mRNA expression at ZT13 in the unfractionated BM of control Nrtn+/+ and compound Nrtn−/− mice. (L) Frequencies of donor-derived WT lin−sca1+ckit+ HSPCs that homed to the BM at ZT5, 6 hours after IV transplantation into nonirradiated Gfra2−/−, Nrtn−/−, and WT mice (at ZT23). (M) Blood CFU-C fold change at ZT5 in WT mice treated with saline, β-AR antagonists, or cholinergic nicotinic (Chrn) antagonist at ZT23. Vcam1 (N) and Sele (O) mRNA expression at ZT5 in the BM of WT mice treated with saline, β-AR antagonists, or cholinergic nicotinic antagonist at ZT23. (C,F-O) Data are mean ± standard error of the mean; n (inside bars) and P values (multivariate analysis for >2 groups) are indicated. *P < .05, **P < .01, ***P < .001, unpaired 2-tailed t test (C,F,J-K), 1-way analysis of variance with Bonferroni comparisons (G-I,L-O). ns, not significant.
We used Nrtn−/− mice to study the influence of local cholinergic signals on vascular adhesion. Previous findings have shown that cholinergic signaling through nicotinic receptor α7 (Chrnα7) activation in endothelial cells reduces Vcam1, E-selectin, and inflammation in the Schwartzman reaction model.49 Interestingly, and mirroring Gfra2−/− mice, Nrtn−/− mice exhibited fourfold reduced Vcam1 and Sele mRNA expression (Figure 7J-K), suggesting that bone sympathetic cholinergic fibers inhibit vascular adhesion.
Expression of adhesion molecules follows day/night oscillations (Figure 3E-G), dropping in the morning. Because bone noradrenergic and cholinergic fibers share a sympathetic origin, we hypothesized that light-induced SNS activity might locally inhibit vascular adhesion and BM homing through cholinergic axons to facilitate noradrenergic-mediated release of HSPCs and leukocytes. We measured diurnal HSPC BM homing in control and Gfra2−/− mice (which lack sympathetic cholinergic innervation) transplanted with labeled cells 1 hour before light onset (ZT23). Importantly, diurnal HSPC homing was increased 40% in Gfra2−/− BM but was reduced 25% in Nrtn−/− BM (Figure 7L). Therefore, whereas β2-adrenergic signals promote nocturnal BM homing, sympathetic cholinergic signals inhibit BM homing at dawn.
These data suggested that sympathetic noradrenergic and cholinergic signals favor diurnal BM egress of HSPCs and leukocytes through different mechanisms: whereas noradrenergic signals decrease BM Cxcl12 expression, cholinergic signals might inhibit vascular adhesion. For confirmation, we injected WT mice with antagonists of β2-AR or β3-AR or cholinergic nicotinic receptors 1 hour before light onset (ZT23), and measured circulating HSPCs and BM adhesion molecule expression 6 hours later (ZT5). In contrast with β2-AR’s role in nocturnal BM homing, β2-AR blockade did not alter circulating HSPCs or adhesion molecules during the morning phase (Figure 7M-O). Therefore, BM β2-AR does not seem to be responsive in the morning, likely as a result of well-known β2-AR desensitization; this might follow sustained stimulation by high nocturnal plasma epinephrine, which preferentially binds β2-AR. In contrast, β3-AR blockade decreased BM egress of HSPCs in the morning(Figure 7M), consistent with our previous study.6 Nicotinic receptor blockade increased Vcam1 and Sele mRNA BM expression 6 hours later (ZT5, morning) (Figure 7N-O), confirming that cholinergic nicotinic signals repress BM homing by inhibiting vascular adhesion in the morning. Interestingly, β3-AR blockade had a similar effect on adhesion molecule expression, suggesting the possible inhibition of vascular adhesion by β3-adrenergic signaling at dawn.
Altogether, our results show that sympathetic cholinergic signals instruct differential responses by BM β-ARs. Sympathetic cholinergic signals cause reduced β3-AR BM expression at night, when higher endocrine-derived epinephrine promotes β2-AR–dependent vascular adhesion and BM homing. This regulation adds to central parasympathetic inhibition of sympathetic noradrenergic tone to promote nocturnal BM homing. In contrast, light-triggered BM sympathetic activity facilitates HSPC and leukocyte egress to the bloodstream through 2 neurotransmitters: norepinephrine reduces Cxcl12-dependent chemotaxis through β3-AR,6 and acetylcholine inhibits vascular adhesion and BM homing (this study).
Discussion
Previous studies have demonstrated that sympathetic noradrenergic fibers regulate HSPC and leukocyte migration,50 but the underlying mechanisms were not fully dissected. Consequently, similar noradrenergic signals were proposed to trigger opposite migratory behaviors of murine HSPCs/leukocytes at different day/night hours: BM egress during the daytime6 (resting period) and nocturnal BM homing (activity period).12 Moreover, these studies assumed that all sympathetic fibers innervating bone are noradrenergic. However, some of these fibers acquire cholinergic properties postnatally,51 but their function has remained unknown.2-4 Although central cholinergic signals have been recently proposed to regulate G-CSF–induced HSC mobilization,11 it was unknown whether the cholinergic nervous system regulates physiological HSC traffic.
In this study, we uncover a dual cholinergic regulation that cooperates with noradrenergic signals to control day/night traffic of HSPCs and leukocytes. These cholinergic signals involve central parasympathetic and peripheral sympathetic innervation, which coordinately act to orchestrate physiological migration of HSPCs and leukocytes.
Our results show that central (parasympathetic) and local (sympathetic) cholinergic signals cooperate to restrict BM egress and render β2-AR–mediated homing predominant at night. Central inhibition of sympathetic tone by parasympathetic signals explains reduced BM egress of HSPCs and leukocytes, which is regulated by β3-AR.6 Locally, sympathetic cholinergic signals modulate the adrenergic response of target cells by reducing their β3-AR expression. In contrast, light triggers local sympathetic (noradrenergic and cholinergic) activity. Noradrenergic signaling (norepinephrine) triggers β3-AR-Cxcl12–dependent BM egress.6 Our novel findings demonstrate that sympathetic cholinergic activity facilitates HSC and leukocyte release by reducing BM vascular adhesion and homing.
These data agree with previous studies reporting increased parasympathetic activity in mice toward the day–night interphase,27 explaining reduced central sympathetic noradrenergic tone45 and BM β3-AR activation, and, consequently, decreased nocturnal egress of HSPCs and leukocytes. However, epinephrine is preferentially released by the adrenal glands at night in mice in response to the hypothalamus–pituitary–adrenal axis.52,53 Moreover, norepinephrine oscillations are abolished by sympathectomy54 or under constant light,55 confirming that neuronal norepinephrine is modulated by photic cues. In contrast, epinephrine oscillations are not affected by these interventions, whereas epinephrine is secreted in response to endocrine signals, such as cortisol.53 Importantly, β2-AR and β3-AR show opposite >30-fold higher affinities for epinephrine and norepinephrine, respectively.16 This evidence and our data suggest that nocturnal endocrine–released epinephrine (and not the SNS) triggers β2-AR–dependent BM homing, whereas light-triggered sympathetic activity induces BM release through norepinephrine (via β3-AR) and inhibits vascular adhesion through acetylcholine (sympathetic cholinergic). Moreover, we show that β2-AR and β3-AR expression oscillates daily and peaks at the same time as those neurotransmitters with highest affinity (diurnal β3-AR–norepinephrine; nocturnal β2-AR–epinephrine), suggesting that coordinated regulation of ligand and receptor ensures robust and timely responses.
This evidence is supported by 2 complementary murine models: Gfra2−/− mice and neurturin (a GFRα2 ligand)–deficient mice (supplemental Table 1). The receptor GFRα2 is required for cholinergic neuron survival (parasympathetic or sympathetic).19,23 We found that Gfra2−/− mice19,56 exhibit halved AChE activity and doubled norepinephrine in urine at night, consistent with extensive parasympathetic deficiency causing derepression of nocturnal sympathetic tone. Exacerbated noradrenergic activity in Gfra2−/− mice increases the bidirectional traffic of HSCs and leukocytes (BM egress and homing). Interestingly, whereas abnormal nocturnal egress is normalized in β3-AR–knockout, but not β2-AR–knockout, mice (Figure 5G), increased BM homing is rescued by β2-AR blockade (Figure 4I), further revealing the different roles of the β-ARs. Diurnal homing to the Gfra2−/− microenvironment is also exacerbated by increased vascular adhesion caused by lack of sympathetic cholinergic innervation (Figure 7L). However, because circulating HSPCs and WBCs accumulate in Gfra2−/− mice at night, β3-AR signaling seems to be predominant and enforces BM egress, overriding increased BM homing. Supporting this possibility, Gfra2−/− mice exhibit increased β3-AR mRNA expression at night (Figure 6B).
In contrast, Nrtn−/− mice have preserved central parasympathetic innervation and, consequently, normal noradrenergic activity.22 However, Nrtn−/− mice exhibit doubled sympathetic cholinergic innervation in bone (Figure 7D-F). Therefore, whereas Gfra2−/− mice lack cholinergic innervation, Nrtn−/− mice represent a gain-of-function model for bone sympathetic cholinergic innervation. Indeed, despite some similarities between the 2 mouse models, Gfra2−/− mice (but not Nrtn−/− mice) exhibit retarded growth. Given that GFRα2 and its coreceptor RET can signal in the same cell (cis) or between neighboring cells (trans) during development,57 future studies will address differences between Gfra2−/− and Nrtn−/− mice. Mirroring Gfra2−/− mice, Nrtn−/− mice exhibit reduced expression of adhesion molecules (Figure 7J-K), further supporting the inhibition of vascular adhesion by sympathetic cholinergic fibers through nicotinic receptors, as previously proposed in other models.49,58
In summary, our results indicate that both branches of the autonomic nervous system (parasympathetic and sympathetic noradrenergic/cholinergic) cooperate to orchestrate rhythmic traffic of HSCs and leukocytes. Light triggers BM sympathetic activity, which reduces β3-AR-Cxcl12–dependent BM chemotaxis through norepinephrine, and inhibits vascular adhesion through acetylcholine. Both effects of local sympathetic activity (noradrenergic and cholinergic) permit the release of HSCs and leukocytes into the circulation. Parasympathetic activation toward the day–night interphase antagonizes the SNS. However, high plasma concentration of endocrine-derived epinephrine at night preferentially stimulates β2-AR to increase vascular adhesion and BM homing.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank C. Kapeni, C. Fielding, A. Sánchez-Aguilera, L. Arranz, and other past members of the S.M.-F. group for help; D. Pask, T. Hamilton, the Central Biomedical Services and Cambridge National Institute for Health Research (NIHR)–Biomedical Research Centre (BRC) Cell Phenotyping Hub (University of Cambridge) for technical assistance; and A. R. Green (University of Cambridge) and Y. Kunisaki (Kyushu University) for data discussion.
This work was supported by core support grants from the Wellcome Trust and the Medical Research Council (MRC) to the Cambridge Stem Cell Institute, the Spanish Ministry of Economy and Competitiveness (SAF-2011-30308), the Pro-CNIC Foundation, Severo Ochoa Center of Excellence award SEV-2015-0505 to CNIC, Ramón y Cajal Program grant RYC-2009-04703, Marie Curie Career Integration grant FP7-PEOPLE-2011-RG-294096, ConSEPOC-Comunidad de Madrid S2010/BMD-2542, Red TerCel (Instituto de Salud Carlos III (ISCIII)-Spanish Cell Therapy Network), National Health Institute Blood and Transplant (United Kingdom), Horizon2020 grant ERC-2014-CoG-64765, and a Cancer Research UK Programme Foundation Award to S.M.-F., who was also supported in part by an International Early Career Scientist grant from the Howard Hughes Medical Institute. A.G.-G. received fellowships from the Ramón Areces and LaCaixa Foundations. C.K. was supported by Marie Curie Career Integration grant H2020-MSCA-IF-2015-708411.
Authorship
Contribution: A.G.-G. performed most of the experiments and analyses, prepared figures, and drafted the manuscript; C.K., M.G.-F., O.D., J.V., D.M.-P., J.I., and J.A.B.-G. performed in vivo experiments and tissue analysis; J.Z., J.A.P.-S., J.J.T.-A., T.M., and M.S.A. provided mice and helped with data analysis; S.M.-F. designed the overall study; performed, supervised, and analyzed experiments; prepared figures; and wrote the manuscript; and all authors edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Simón Méndez-Ferrer, Long Rd, Box 96, Cambridge CB2 0PT, United Kingdom; e-mail: sm2116@medshcl.cam.ac.uk.