Abstract
In contrast to other diverse therapies for the X-linked bleeding disorder hemophilia that are currently in clinical development, gene therapy holds the promise of a lasting cure with a single drug administration. Near-to-complete correction of hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) have now been achieved in patients by hepatic in vivo gene transfer. Adeno-associated viral vectors with different viral capsids that have been engineered to express high-level, and in some cases hyperactive, coagulation factors were employed. Patient data support that sustained endogenous production of clotting factor as a result of gene therapy eliminates the need for infusion of coagulation factors (or alternative drugs that promote coagulation), and may therefore ultimately also reduce treatment costs. However, mild liver toxicities have been observed in some patients receiving high vector doses. In some but not all instances, the toxicities correlated with a T-cell response directed against the viral capsid, prompting use of immune suppression. In addition, not all patients can be treated because of preexisting immunity to viral capsids. Nonetheless, studies in animal models of hemophilia suggest that the approach can also be used for immune tolerance induction to prevent or eliminate inhibitory antibodies against coagulation factors. These can form in traditional protein replacement therapy and represent a major complication of treatment. The current review provides a summary and update on advances in clinical gene therapies for hemophilia and its continued development.
Introduction
Hemophilia is an X-linked monogenic coagulation disorder resulting from a deficiency in coagulation factors in the intrinsic coagulation cascade.1,2 Hemophilia A, the more prevalent form of hemophilia, occurs in 1 in 5000 live male births and is caused by a mutation in the gene coding for factor VIII (FVIII), resulting in the loss of functional FVIII protein. FVIII is a critical cofactor for the serine protease factor IX (FIX), which is deficient in patients with hemophilia B. Both FVIII and FIX are naturally synthesized in the liver: FVIII in liver sinusoidal endothelial cells (LSEC) and FIX in hepatocytes. It is estimated that there are a total of 20 000 patients with hemophilia in the United States, with hemophilia A being about 6 times more common than hemophilia B. Clinically, both patients with hemophilia A and patients with hemophilia B are segregated into 3 groups based on residual coagulation factor activity: severe (< 1%), moderate (1%-5%), and mild (5%-40%). Untreated patients with severe hemophilia are at risk for either morbidity or mortality from spontaneous or trauma-induced bleeds. The most common form of morbidity is hemophilic arthropathy resulting from recurrent bleeds into the joints. Patients with moderate hemophilia have a significant reduction in spontaneous bleeds, but are still at risk from trauma-induced bleeds, and patients with mild hemophilia may appear phenotypically normal and never show signs of uncontrolled bleeds unless undergoing severe trauma or surgery.
Current recommended therapy for hemophilia is prophylactic administration of exogenous coagulation factors derived from pooled plasma or recombinant protein. The short biological half-lives of FVIII and FIX proteins require frequent infusions (2-3 times per week) to maintain trough levels above 1%, the minimally effective level to significantly reduce the incidence of spontaneous bleeds. A major complication of factor replacement therapy is the formation of anti-drug antibodies, termed inhibitors.3 Inhibitors form in approximately 25% to 30% of patients with hemophilia A and, less frequently, in 3% to 5% of patients with hemophilia B. Clinically, patients with an inhibitor titer above 5 Bethesda units (1 Bethesda unit is defined as the amount of antibody that reduces factor activity by 50%) are no longer responsive to factor replacement, and require treatment with bypassing agents to maintain hemostasis. Traditional bypassing agents, such as activated prothrombin complex concentrate and recombinant activated FVII, are generally expensive, have short biological half-lives, and are not as effective as FVIII or FIX in long-term hemostasis. Alternatively, inhibitor patients can be placed on an immune tolerance induction (ITI) protocol requiring frequent infusions of super physiological levels of coagulation factor until inhibitors are reduced or eliminated and patients can resume factor replacement therapy.4,5 Although effective in approximately two-thirds of patients with hemophilia A with inhibitors, ITI often has to be discontinued in patients with hemophilia B because of the development of anaphylaxis and nephrotic syndrome.6 ITI therapy is expensive and places a significant burden on the patient, and the long duration of therapy increases the risk for bleeds.7 Considering the high lifetime costs, frequencies of infusions, and potential health burden, there is a need for alternative cost-effective therapies with reduced risk and improved efficacy for hemophilia.
Rationale for gene therapy for hemophilia
Gene therapy provides a functional copy of the disease-causing gene that is either absent or expressed as a nonfunctional protein; thus, it can be highly effective in treating monogenic disease, such as hemophilia. The initial barrier of inefficient delivery of the therapeutic genetic payload into target cells and tissues was circumvented through the adoption of viral vectors derived from mammalian viruses that have naturally evolved to deliver their genetic cargo into cells and tissues. These vectors contain minimal wild-type viral sequences, and their pathogenic, replicative, and structural viral genes are replaced with the therapeutic gene cassette. Over the years, hepatic in vivo gene transfer using adeno-associated viral (AAV) vectors has shown the best success in preclinical and clinical studies, with several clinical studies for both hemophilia A and B enrolling patients for phase 3 trials.
Hemophilia is well suited for correction by gene therapy because the bleeding phenotype is responsive to a wide range of factor levels, and precise regulation is not necessary. Further, as clotting factor proteins are secreted into the circulation, it is possible to correct the bleeding diathesis with gene delivery to a fraction of total hepatocytes. FVIII and FIX can be synthesized in nonnative cells and tissues. For example, even though FVIII is naturally secreted by specialized endothelials such as LSEC and extrahepatic endothelial cells, expression in hepatocytes generates a functional protein that has restored hemostasis in animal models and human patients. And finally, for patients in developing countries with comorbidities and mortalities resulting from inadequate factor supply, gene therapy could provide a significant benefit by providing a continuous source of clotting factor from a single treatment.
AAV vectors
AAV vectors are derived from wild-type AAV,8 a member of the parvovirus family. Wild-type AAV is nonpathogenic, weakly immunogenic, and replication deficient, requiring a helper virus for replication. AAV vectors can deliver a therapeutic transgene cassette up to 5 kb into both dividing and nondividing cells. However, the majority of AAV vector genomes do not integrate into the host genome, and gene delivery into dividing cells results in progressive loss of transgene. AAV serotypes are segregated into clades based on viral capsid homology.9 The viral capsid governs both tissue tropism and intracellular viral particle trafficking. Engineered AAV capsids generated by random and directed mutagenesis and capsid gene shuffling have been created to enhance human hepatocyte tropism, resist neutralizing antibodies, and evade elimination of transduced hepatocytes by cytotoxic T cells.10-12 Engineering of the expression cassette continues to be a key component for successful gene therapy. For instance, vector potency can be enhanced through the design of stronger synthetic liver-specific promoters, codon-optimized F8 and F9 cDNAs, and use of engineered F8 (B-domain deleted FVIII variants) and F9 (hyperactive FIX-Padua variant R338L) genes.13-19 At the same time, there is concern that overexpression, in particular of FVIII, could result in cellular stress and unwanted toxicities.20-22 Some clinical trials use expression cassettes that have been edited to eliminate immune stimulatory CpG motifs, which have the potential to enhance CD8+ T-cell responses.14,23 However, as the inverted terminal repeats of the viral genome also contain CpG motifs, their complete elimination from the vector is not possible.
AAV vector liver directed gene therapy in hemophilia animal models induces immune tolerance to FVIII and FIX through induction of factor-specific regulatory T cells (Treg).24-29 There is now strong evidence that AAV and lentiviral (LV) vector liver directed gene therapy can also eliminate established inhibitors.30 For instance, AAV-F8 and AAV-F9 vector liver gene transfer was an effective ITI in canine hemophilia models.27,28 Similarly, LV-F931 and AAV-F926 vector liver gene transfer could eradicate pathogenic inhibitors in hemophilia B mice. Although these results are encouraging, translatability to humans will have to be confirmed.
Immunology of AAV gene therapy
Immune recognition by cytotoxic CD8+ T cells or antibody responses to the vector capsid, the transgene product, or both can compromise therapeutic expression of the transgene (Figure 1).32-34 However, an immune response that is detectable by in vitro assays does not always affect transgene expression in vivo.18,35 Humans are naturally infected with wild-type AAV during childhood, and thus may develop neutralizing antibodies (NABs) that often cross-react with multiple serotypes and prevent gene transfer with AAV vectors. Similarly, NAB development on gene transfer prevents patients from receiving repeat administration of the same AAV vector, at least in the near future. Activation of capsid-specific CD8+ T cells may result in targeting of transduced hepatocytes, mild and transient transaminitis, and loss in factor levels.35 Hence, an immune suppression protocol was developed based on steroid drugs that are given in response to increases in transaminases in the blood.14 Rapid employment is required to preserve transgene expression. Compared with T-cell assays, elevation in transaminases allows for more rapid treatment response. Although there is evidence for memory CD8+ T cells to capsid, this response is not fully understood in humans, and loss of transgene expression or hepatotoxicity could not always be correlated with a T-cell response.23,35,36 Therefore, a better understanding of the innate and adaptive immune responses to AAV vectors, as well as alternative sources of toxicity, is needed to ensure long-term gene expression.11,37,38
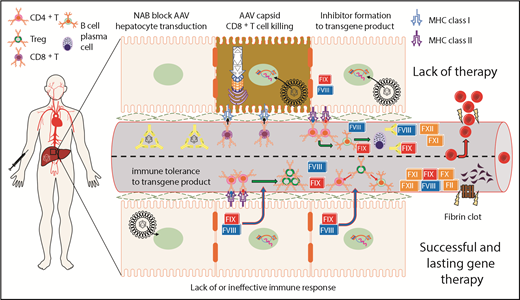
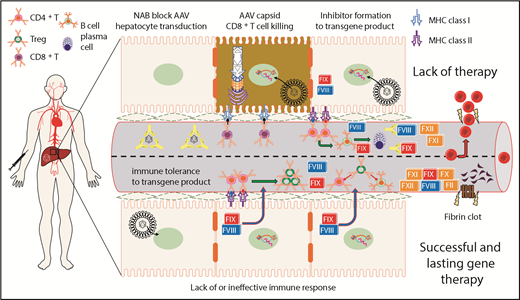
Summary of immunological challenges and successes of liver-directed AAV gene therapy for hemophilia in humans. This is a representation of hepatocytes and LSEC-lined blood vessel within the liver. After intravenous delivery of an AAV vector, viral particles enter the hepatocyte, escape the endosome, and deliver their genetic payload to the nucleus. Preclinical animal models show that FVIII or FIX expression in the liver induces immunological tolerance through conversion of effector CD4+ T cells into Treg. Treg suppress activation of both B- and T-cell responses directed against FVIII or FIX. Preexisting NAB to the AAV capsid can block hepatocyte gene delivery and prevent therapeutic correction of the bleeding diathesis. On successful gene delivery to hepatocytes, primary or recall immune responses may lead to activation of capsid-specific cytotoxic CD8+ T cells and targeted elimination of hepatocytes presenting AAV capsid epitopes on MHC class I. Preclinical animal models of severe hemophilia show that suboptimal gene delivery may fail to induce tolerance to the transgene product because a threshold level of FVIII or FIX expression is needed for successful activation of Treg. In the absence of Treg, effector CD4+ T cells may become activated on recognition of FVIII or FIX epitopes presented on MHC class II, which is primarily expressed on professional antigen presenting cells. The resulting T help promotes B-cell maturation, leading to the production of high-affinity class-switched antibodies and terminal differentiation into antibody secreting plasma cells.
Summary of immunological challenges and successes of liver-directed AAV gene therapy for hemophilia in humans. This is a representation of hepatocytes and LSEC-lined blood vessel within the liver. After intravenous delivery of an AAV vector, viral particles enter the hepatocyte, escape the endosome, and deliver their genetic payload to the nucleus. Preclinical animal models show that FVIII or FIX expression in the liver induces immunological tolerance through conversion of effector CD4+ T cells into Treg. Treg suppress activation of both B- and T-cell responses directed against FVIII or FIX. Preexisting NAB to the AAV capsid can block hepatocyte gene delivery and prevent therapeutic correction of the bleeding diathesis. On successful gene delivery to hepatocytes, primary or recall immune responses may lead to activation of capsid-specific cytotoxic CD8+ T cells and targeted elimination of hepatocytes presenting AAV capsid epitopes on MHC class I. Preclinical animal models of severe hemophilia show that suboptimal gene delivery may fail to induce tolerance to the transgene product because a threshold level of FVIII or FIX expression is needed for successful activation of Treg. In the absence of Treg, effector CD4+ T cells may become activated on recognition of FVIII or FIX epitopes presented on MHC class II, which is primarily expressed on professional antigen presenting cells. The resulting T help promotes B-cell maturation, leading to the production of high-affinity class-switched antibodies and terminal differentiation into antibody secreting plasma cells.
Vector dose and transgene expression levels have a significant effect on whether an immune response is induced against both the AAV vector and transgene product.35,38 Importantly, optimal transgene expression in the hepatic environment can lead to immune tolerance to the therapeutic protein antigen, and to date, no clinical gene therapy study has reported detecting B- or T-cell response directed against either FVIII or FIX.14,23,36,39-41 Initial clinical studies for AAV gene therapy for hemophilia have been conducted in adults older than 18 years, in which patients have had sufficient exposure days without an inhibitor, a common exclusion criterion. Additional rigorous clinical studies will be needed in previously untreated patients, more so for patients with hemophilia A, in whom the inhibitor frequency is 25% to 30%, to determine the relative risk for inhibitor formation after gene transfer. Further, as the expression cassettes for FVIII and FIX are episomal, treating a patient too early, while the liver is still growing, may cause a loss in expression. If a patient would require additional vector infusions, strategies to prevent the formation of neutralizing antibodies against the vector capsid will have to be developed, or a vector with diverse capsid sequence will have to be used for the second administration.
AAV gene therapy clinical trials
AAV vectors have progressed into clinical studies for both hemophilia A and B. Gene transfer of a scAAV8-coF9 vector to human hepatocytes in adult patients with hemophilia B has provided stable expression of FIX protein now for more than 6 years.40 The ClinicalTrials.gov database presently lists 9 active clinical trials evaluating different AAV capsid serotypes and F9 gene sequences for hemophilia B and 9 trials to evaluate AAV-F8 gene therapy for hemophilia A (Table 1), with some overlap, as the same vector is being evaluated in both phase 1/2 and 3 studies. Although ongoing clinical trials for hemophilia have been recently reviewed elsewhere,42-48 this review will focus on several notable trials that have recently been published or presented updates at national meetings for the American Society of Hematology and American Society of Cell and Gene Therapy.
Hemophilia A clinical trials
BioMarin initiated the first clinical trial for hemophilia A, using liver gene transfer of an AAV5 vector expressing a codon optimized BDD-FVIII-SQ protein, BMN 270.36 An initial patient treated at a low vector dose had nearly undetectable FVIII levels with little change in FVIII protein usage. A second patient treated at an approximately 3-fold higher dose had stable FVIII levels, between 1% to 3% normal, and discontinued prophylactic FVIII protein usage. Patients in the highest-dose cohort (reported as 6 × 1013 vector genomes [vg]/kg) expressed a range of therapeutic FVIII levels, with median of 77% and range of 19% to 164% of normal at 52 weeks, with a significant reduction in recombinant FVIII usage and annual bleeding rate. Most patients treated in the high-dose cohort received an extended course of prophylactic prednisolone in response to mild elevations in transaminase observed in participant 3. Participant 4 developed transaminitis after cessation of the initial round of steroid treatment and was placed back on a second round of tapered steroids. Elevation in transaminases did not correlate to a T-cell response, prompting the authors to speculate that vector infusion itself combined with innate sensing of viral molecular structures may have caused mild liver inflammation.36 A step-down dose cohort with 6 participants received 2/3 of the highest dose. A median expression of 33% of normal at 24 weeks postvector treatment and discontinuation of FVIII protein therapy with no reported bleeds was documented. BioMarin reported at the 2018 World Federation of Hemophilia congress some reduction in the median FVIII levels at 104 weeks, with follow-up studies forthcoming.
BMN 270 was produced using a baculoviral insect cell-based production system. This was the first hemophilia trial to use rapid vector dose escalation (vector dose was raised after the first treated patient failed to show any correction) and prophylactic prednisolone administration. BMN 270 was recently assigned the commercial name of Valoctocogene Roxaparvovec. Two phase 3 studies with an estimated enrollment of 130 patients total have been initiated with target vector dose of 4 × 1013 vg/kg and 6 × 1013 vg/kg. In addition, BioMarin is enrolling by invitation a phase 1/2 study to evaluate high-dose Valoctocogene Roxaparvovec gene delivery in patients with hemophilia A with preexisting AAV5 capsid antibodies.
Spark Therapeutics has presented preliminary trial data on patients with hemophilia A treated with an initial low vector dose in 2 patients, followed by 3 patients treated with intermediate dose, and the 7 treated with high dose of their Spk8011 vector (representing 2-fold dose escalations between cohorts).49-51 The reported vector doses ranged from 5 × 1011 to 2 × 1012 vg/kg. Although these numbers indicate usage of substantially lower vector doses compared with the trial with AAV5, titers were not determined side by side and were likely not based on identical protocols. The first 2 patients at the initial vector expressed 10% and 13% normal levels of FVIII protein, and 2 of the 3 mid-dose–treated patients expressed 8% and 12% of normal FVIII protein. According to data released by Spark Therapeutics, higher levels of expression (on average, ∼30% and up to 49% of normal FVIII activity by 3 months), but also toxicities, were observed at the highest vector dose. This trial is notable, as it is the first use of capsid that was generated from a capsid library after selection for human hepatocyte tropism and resistance to antibody neutralization.10
Hemophilia B clinical trials
Of the many ongoing hemophilia B AAV clinical trials, there are several notable successes. The trial sponsored by University College, London (UCL) and St. Jude Children’s Research Hospital for patients with hemophilia B, using an scAAV2/8-LP1-hFIXco vector, was the first long-term success story for hemophilia gene therapy.40 The first patient was treated in 2009, making this trial the longest follow-up on the safety and stability of FIX protein expression from an AAV vector in the liver. However, several patients had an early decline in FIX levels related to transient transaminitis associated with a capsid T-cell response that stabilized after treatment with tapered prednisolone. Last reported FIX levels in the high-dose cohort (2 × 1012 vg/kg) of 6 patients were a mean of 6.1% with a range of 2.89% to 7.2%. Going forward, the UCL has initiated a new phase 1/2 trial for hemophilia B, using an undisclosed (capsid and transgene) AAV-F9 vector, FLT180a. As of this review, no data have been released.
Spark Therapeutics has initiated a hemophilia B clinical trial, using an AAV8 capsid with several amino acid substitutions to package a hyperactive FIX-Padua expression cassette.39 FIX-Padua is a naturally occurring hyperactive FIX variant (R338L) with a reported approximately 8-fold increase in specific activity.52 Ten patients were treated at a dose of 5 × 1011 vg/kg and had a mean FIX activity of 33.7% with a range of 14% to 81%. Two participants, 7 and 9, had asymptomatic transient transaminitis that was resolved through steroid treatment, and only 1 subject showed moderate loss in FIX activity. Eight of the 10 treated patients had stable FIX activity during reported follow-up, and no indication of an AAV capsid T-cell response. At this time, new patients are being enrolled for a phase 3 trial.
UniQure has recently published data on their clinical trial for hemophilia B with an AAV5 vector using the same F9 expression cassette as the UCL and St. Jude Children’s Research Hospital trial.14,23,40 Ten patients were enrolled, and 5 received a low dose; another 5 were treated at a 4-fold higher dose. Mean FIX levels in the low-dose cohort were 4.4% with a range of 1.3% to 6.8%. Mean FIX in the higher-dose cohort were 6.9% with a range of 3.1% to 12.7%. However, it appears that both hemophilia B trials by UCL and Spark used lower vector doses to achieve similar or higher efficacy. Similar to the BioMarin trial, several patients displayed transient transaminitis without indication of a capsid T-cell response. Given the high vector doses used, it may be of interest to examine other potential causes for hepatotoxicity in these vector preparations.53 A new dose confirmation trial using the FIX-Padua variant at the high dose (reported as 2 × 1013 vg/kg) is planned and expected to enroll 3 patients with hemophilia B, and depending on outcomes, will proceed to a phase 3 study.
There are several hemophilia B AAV liver gene therapy trials that have been completed or terminated on the ClinicalTrials.gov database, primarily because of the lack of efficacy. However, most of these trials have never published patient outcomes in a peer-reviewed journal, so there are limited publicly available data.35,54 A common reporting of most of these studies is transient transaminitis in either the presence or absence of detectable capsid-specific CD8+ T cells.
Challenges to clinical implementation of AAV gene therapy
As discussed earlier, there are a number of immunological hurdles to AAV-mediated gene therapy for hemophilia. Additional challenges are summarized here.
Production scale-up of AAV vectors
For widespread therapeutic use, suitable scalable production methods will be needed to produce sufficient quantities of clinical-grade AAV vectors. Either transfection of mammalian cells, such as HEK293 cells, or baculoviral production systems in insect cells have been used to manufacture AAV vectors in all hemophilia clinical trials to date.18 Although further improvements are being made to the baculoviral system, a vaccinia virus-based platform has more recently been developed for scale up in mammalian cells.55,56 Permanent production cell lines for AAV are also under development. The potential role of the production system in immune responses and toxicities remains to be defined.
Nonimmune toxicities related to gene transfer
The risk for insertional mutagenesis after AAV-mediated gene transfer is low, as the majority of vector genomes persist episomally. Even though natural infection with AAV is not linked to cancer, deep sequencing studies show that integration of the AAV genome can occur in the liver.57,58 In addition, an increased incidence of hepatocellular carcinoma (HCC) has been reported in the mucopolysaccharidoses type VII mouse model after perinatal gene transfer of AAV, potentially through integration and disruption of an imprinted region rich in miRNAs and snoRNAs on mouse chromosome 12.59 Subsequent studies in other murine models have failed to recapitulate this finding. However, an extensive study in mice revealed that vector design can modulate the relative risk for HCC development.60 Collectively, the available data in various animal models suggest that AAV has a relatively low risk for tumorigenesis,61 although AAV integrations appear to be present in some human HCC.62 There are several recent studies demonstrating links to HCC and wild-type AAV.63,64 A debate whether this extends to AAV vectors is ongoing.65-67 Although there is substantially more evidence supporting no risk for insertional mutagenesis in animal models and AAV-treated patients with hemophilia, greater numbers of treated patients are still needed to completely rule out this as a potential risk.18
Another potential toxicity, related specifically to AAV gene therapy for hemophilia A, has been described by recent research. This work demonstrated that overexpression of FVIII in hepatocytes may induce cellular stress20,21 as a result of the accumulation of misfolded FVIII protein in the endoplasmic reticulum (ER). Because hepatocytes do not naturally express FVIII protein, nor the closely associated vWF protein, the risk for ER-induced stress in hepatocytes is a potential concern. However, it is not clear that an ER stress response to FVIII occurred in AAV-treated patients with hemophilia A, as discussed by the investigators.36 Nonetheless, preclinical studies have focused on engineering FVIII proteins that are more efficiently secreted from hepatocytes,15,19,67-71 including the FVIII variant FVIII-V3, which is now being evaluated in clinical trial (Table 1). In addition to ER stress, it was speculated that the mild liver toxicities observed in patients with hemophilia A may be related to viral particle trafficking, uncoating, and vector DNA induction of the DNA damage response.36
Evolving approaches for hemophilia gene therapy
Gene editing and targeted integration
Gene editing is a molecular technique that can correct the endogenous genetic defect or direct the integration site of a therapeutic gene through nuclease-targeted double-stand DNA breaks and homology-directed repair. Zinc-finger nucleases, transcription activator-like effector-based nucleases, and the clustered regularly interspaced short palindromic repeats-Cas systems were developed to induce a double-strand break in DNA, vastly increasing the frequency of homology-directed repair and editing, or for gene disruption.72,73 In terms of hemophilia therapeutics, gene editing approaches are being investigated to correct endogenous F9 mutations or insert F9 into a defined locus such as safe harbor sites or, more recently, the albumin locus.74 Clinical application may be limited by off-target cutting and higher-than-anticipated frequency of integrations with the nonhomologous end joining pathway. Sangamo Therapeutics is currently recruiting patients using AAV6 to deliver zinc-finger nucleases and a promoterless F9 transgene, targeting the albumin locus, to treat hemophilia B. However, at the time of this review, there have been no data released. Alternatively, it was shown that an AAV cassette with homology arms is sufficient for targeted integration independent of DSB in mice resulting in FIX levels in the range of 7% to 20% normal, thus avoiding the need for codelivery of an endonuclease.75
Combining gene and immune modulatory therapies
Although hepatic gene transfer has the potential to induce immune tolerance, adjunct immune modulation may further enforce tolerance to the FVIII of FIX transgene products. This may be of particular relevance for the more immunogenic FVIII molecule. A number of such combination therapies have been described. The mammalian target of rapamycin inhibitor rapamycin has received particular attention, as it causes activated induced cell death after signaling through the T-cell receptor in CD4+ T effector cells while sparing Treg. In murine models, a 1-month regimen based on administration of rapamycin and FVIII or FIX antigen (in some cases combined with a second drug with synergistic effects) has been successful in promoting tolerance in murine models of hemophilia in the context of protein or gene therapy.76-78 Nanoparticle delivery of rapamycin is effective at suppressing AAV cellular and humoral immunity.79 Transient B-cell depletion by monoclonal antibody therapy (using anti-CD20) reduced the risk for inhibitor formation in the context of AAV gene therapy,80 and when combined with rapamycin, enhanced reversal of FVIII inhibitors in hemophilia A mice.81 A similar protocol is in clinical development to prevent antidrug antibodies in other enzyme replacement therapies and to suppress NAB formation against AAV vectors, thus improving the chances for successful readministration of vector.82 Of note, there are also nonimmune suppressive methods to promote vector administration in an individual with preexisting NAB to AAV, such as coadministration of decoy capsids (“empty” capsids that lack vector genomes) in excess to saturate NAB binding sites.83
Conclusions
Given the exciting breakthroughs with new hemophilia therapies, there is debate regarding the benefits of gene therapy, considering recently approved enhanced half-life factors and other biologics. However, enhanced half-life products thus far have failed to show a substantial gain in half-life for FVIII. Biologics that target coagulation pathway inhibitory proteins and a bispecific antibody FVIII mimetic show much promise (but have also been associated with thrombosis in clinical evaluation, perhaps because of a lack of functional regulation that is inherent in FVIII and FIX products).48,84 Alternative gene therapy approaches targeting hematopoietic stem cells with lineage-restricted expression in CD68 monocytes71 and platelets85 are being investigated. Targeting expression to platelets may avoid neutralization of FVIII by inhibitors, and also has potential for immune tolerance induction. However, these approaches may require preconditioning of bone marrow at least with a nonmyeloablative regimen and transient immune suppression.
Two separate AAV-based gene therapy phase 1/2 clinical trials for hemophilia A have now reported stable FVIII levels after a single vector dose, demonstrating that gene therapy is the most effective investigational treatment of patients with adult hemophilia A. In hemophilia B, even though enhanced half-life FIX products have benefited from a greater improvement in half-life, multiple phase 1/2 gene therapy, clinical trials have now reported stable therapeutic FIX expression after a single treatment using an AAV vector. Given the robustness of expression thus far, up to 6 years, it is likely that AAV gene therapy for hemophilia B will be a highly attractive therapeutic option for patients after the anticipated approval by the US Food and Drug Administration. Although the clinical data supporting gene therapy for hemophilia have been extremely positive, the enrolled subjects have a preselection bias, and thus some caution is advised in extending the benefits of gene therapy to the general population of patients with hemophilia. It is unclear how effective AAV gene therapy will be in young children with fewer than 50 exposure days, in terms of the risk for inhibitor and potential loss of factor expression over time as a result of dilution of AAV-transduced hepatocytes during liver growth. Integrating LV may provide an alternative vector platform for treating children with hemophilia with in vivo liver gene delivery of immune stealth and tolerogenic LV vectors to hepatocytes86,87 and LSEC.88 In children and adults with either high-risk mutations for inhibitors, history of inhibitor, or presently with inhibitor, it is unclear whether the robust AAV mediated liver tolerance and ITI observed in hemophilia animal models will translate in humans. Therefore, until such studies have been conducted, there is also a need for the continued development of improved factor and bypassing agent products. Nonetheless, with continued growth and success, AAV gene therapy may replace enzyme replacement therapy as a potential curative therapy for hemophilia.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases grant R01 AI51390 and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL097088 and R01 HL131093.
Conflict-of-interest disclosure: R.W.H. received royalty payments from Spark Therapeutics for license of AAV gene transfer technology and serves on a scientific advisory board for Applied Genetic Technologies Corporation. The remaining authors declare no competing financial interests.
Authorship
Contribution: The authors each contributed to the writing of this manuscript.
Correspondence: Roland W. Herzog, IUPUI-Wells Center for Pediatric Research, 1044 W Walnut St, Indianapolis, IN 46202; e-mail: rwherzog@iu.edu; and David M. Markusic, IUPUI-Wells Center for Pediatric Research, 1044 W Walnut Street, Indianapolis, IN 46202; e-mail: dmarkusi@iu.edu.