Key Points
PfGARP is a secreted protein that binds to human erythrocyte band 3.
PfGARP induces RBC aggregates in vitro and may serve as a biomarker of disease progression.

Abstract
Malaria remains a major global threat to human health and economic development. Microvascular lesions caused by Plasmodium falciparum–infected human erythrocytes/red blood cells are hallmarks of severe pathogenesis contributing to high mortality, particularly in children from sub-Saharan Africa. In this study, we used a phage display complementary DNA library screening strategy to identify P falciparum glutamic acid–rich protein (PfGARP) as a secreted ligand that recognizes an ectodomain of human erythrocyte anion-exchanger, band 3/AE1, as a host receptor. Domain mapping of PfGARP revealed distinct nonoverlapping repeats encoding the immune response epitopes and core erythrocyte-binding activity. Synthetic peptides derived from the erythrocyte-binding repeats of PfGARP induced erythrocyte aggregation reminiscent of the rosetting phenomenon. Using peptides derived from the immunogenic repeats, a quantitative immunoassay was developed to detect a selective immune response against PfGARP in human plasma samples obtained from patients in rural Mali, suggesting the feasibility of PfGARP as a potential biomarker of disease progression. Collectively, our results suggest that PfGARP may play a functional role in enhancing the adhesive properties of human erythrocytes by engaging band 3 as a host receptor. We propose that immunological and pharmacological inhibition of PfGARP may unveil new therapeutic options for mitigating lesions in cerebral and pregnancy-associated malaria.
Introduction
Malaria is a devastating parasitic disease that afflicts a massive number of people globally.1,2 According to the World Health Organization, an estimated 216 million cases of malaria infection were reported worldwide, resulting in 445 000 deaths, mainly children in sub-Saharan Africa.3 The parasite induces extensive modifications of host erythrocytes by exporting hundreds of proteins via complex trafficking mechanisms. These proteins are usually expressed at the trophozoite and schizont stages of parasite development, and they play a unique role in Plasmodium falciparum surface virulence and survival resulting from enhanced adhesiveness of infected erythrocytes/red blood cells (iRBCs).4-6 Erythrocyte modifications by malaria-exported proteins are believed to affect red blood cell (RBC) sequestration, a strategy used by the parasite to avoid immune surveillance and splenic clearance of iRBCs in the peripheral blood. Increased stickiness of iRBCs and, to a lesser extent, uninfected RBCs (uRBCs), is one of the main factors contributing to the virulence of P falciparum.7,8 Generally, the adhesive properties are classified into binding of iRBCs to endothelial receptors (cytoadherence), iRBCs to uRBCs (rosetting), iRBCs to other iRBCs (autoagglutination), iRBCs to platelets (platelet-mediated clumping),1 and uRBCs to each other (RBCs aggregation).7,9 Despite recent progress, relatively little is known about the role of hundreds of undefined malaria ligands and their corresponding host receptors in the cellular adhesion pathways.
Phage display complementary DNA (cDNA) technology is a useful tool for the identification of potential ligand–receptor interactions relevant to drug discovery and vaccine development.10-14 We and other investigators have successfully used this technology to identify novel ligand–receptor interactions at the blood stage of malaria infection.15-18 In this study, we screened multiple P falciparum phage display cDNA libraries using human RBCs as bait and identified P falciparum glutamic acid–rich protein (PfGARP) as a potential ligand that binds to the surface of intact RBCs. PfGARP (PF3D7_0113000, PFA0620c), 26% of which is made up of glutamic acid residues,19 is one of the exported proteins with a Plasmodium export element motif that targets hundreds of malaria proteins to specific sites within the host RBCs.6,20 Although the function of PfGARP is not yet known, it is expressed at the trophozoite and schizont stages of parasite development concomitant with the cytoadherence phenomenon.6,21,22 Antibodies against PfGARP have been detected in the plasma of children resistant to malaria.23 The PfGARP homologs have been detected in different isolates of P falciparum; however, this gene is not found in other species of human malaria parasites, including P vivax.6,19 Recent studies indicated the existence of PfGARP in the primate parasites including P reichenowi and P gaboni, which are closely related to the human malaria parasite P falciparum.24-26 These observations suggest that PfGARP may play a functional role that is unique to the lesions caused by P falciparum. PfGARP gene expression and protein expression were increased in parasites isolated from Tanzanian children with malaria,23 suggesting a potential role for PfGARP in promoting severe episodes of malaria pathogenesis.
PfGARP contains 3 distinct lysine-rich repeats, amino acids (AAs) 119 to 163, 253 to 340, and 372 to 446, which are responsible for the localization of the protein to the RBC membrane, as detected by the translocation of GFP-tagged fusion proteins in P falciparum 3D7 parasites.27 Here, we have identified specific repeats of PfGARP that directly bind to human RBCs via an extracellular segment of host band 3 and evaluated a potential role for PfGARP in erythrocyte-adhesion phenomena. We also developed a new monoclonal antibody (mAb) against a distinct region of PfGARP repeats. By combining the mAb and peptide-based scanning of PfGARP repeats, an immunoassay was developed for monitoring the pathogenicity of malaria infection in endemic areas. Functional implications of the newly identified host–parasite interface involving PfGARP and host band 3 will be discussed.
Materials and methods
Phage display cDNA library screens
Phage display cDNA screens were performed to identify human RBC-binding proteins of P falciparum, as previously described.18 Phage cDNA libraries were constructed from P falciparum 3D7 messenger RNA using the T7 select system (Novagen), as described.18 Phage clones were selected through 4 rounds of biopanning using human RBCs as bait. To identify potential host RBC receptors for PfGARP, reverse phage display screens were performed using purified recombinant PfGARP. Using Novagen’s T7 select system, multiple cDNA libraries were constructed from human reticulocyte messenger RNA with random-primed or oligo(dT)-primed cDNA synthesis protocols. Details about the phage display screens can be found in supplemental Materials and methods (available on the Blood Web site).
Expression and purification of PfGARP constructs
Details about the PfGARP constructs, their expression, and purification are included in supplemental Materials and methods and Additional results.
Enzyme treatment of human RBCs and binding assays
Human blood was collected from healthy deidentified donors of both sexes in acid-citrate-dextrose buffer with approval under IRB #9750. Alternatively, human RBCs (AB+) from deidentified donors of both sexes were obtained as packed cells from the American Red Cross (Dedham, MA). The glycophorin B (GYPB)-null RBCs were obtained from ITxM Clinical Services (Rosemont IL). Further details about the enzyme treatment of RBCs and binding assays are included in supplemental Materials and methods.
CHO-K1 erythrocyte–binding assay
CHO-K1 cells (Sigma) were cultured in Ham’s F-12 modified medium supplemented with 10% HyClone fetal bovine serum, 1× l-glutamine, and 1% penicillin-streptomycin. Cells were harvested with 0.05% trypsin-EDTA, and RBC-binding assays were performed essentially as described previously.28-30 Further details about the binding of CHO-K1 cells to RBCs can be found in supplemental Materials and methods .
Human erythrocyte–aggregation assay
Blot overlay assay (far western blotting)
Untreated and enzyme-treated RBC ghosts were prepared from 100 µL of packed human RBCs using cold lysis buffer.32,33 A blot overlay assay was performed as described previously with slight modifications.34 Additional details about the blot overlay assay are included in supplemental Materials and methods.
Pull-down assay
Malaria parasite culture and invasion assay
P falciparum 3D7 parasites were cultured as described previously.37-39 To harvest parasite culture supernatant, P falciparum 3D7 schizont-infected RBCs were isolated using LS magnetic columns (Miltenyi Biotec). Additional details are included in supplemental Figure 2.
Generation of a mAb against PfGARP
Immunization of mice with PfGARP-M
Six BALB/c female mice (8-12 weeks old) were immunized with PfGARP-M protein. Recombinant TRX–PfGARP-M protein was expressed in Escherichia coli BL21 (DE3) and purified using Ni-beads affinity chromatography. Eluted protein was further purified by Mono Q ion exchange chromatography using an AKTA FPLC system (GE Healthcare). Additional details about mAb generation can be found in supplemental Materials and methods.
Characterization of GM7 mAb
Hybridoma clone #7 was selected from 32 positive hybridomas for further characterization. Hybridoma clone #7 was further subcloned by limiting dilution and secreted an immunoglobulin G3 mAb termed GM7-1 (for simplicity, we call it GM7 monoclonal). Further details about mAb characterization are included in supplemental Materials and methods.
Development of an ELISA for PfGARP
Streptavidin-coated plates, as well as noncoated enzyme-linked immunosorbent assay (ELISA) plates, were used to immobilize His–PfGARP-M antigen or synthetic peptides of interest by incubation overnight at 4°C. Additional information about ELISA development and synthetic peptides can be found in supplemental Materials and methods.
Localization of PfGARP by immunofluorescence microscopy
A thin blood smear of P falciparum 3D7 culture was air-dried, fixed, and permeabilized with ice-cold methanol for 45 minutes. Slides were washed briefly with 0.05% TWEEN 20 in phosphate buffered saline. GM7 mAb was diluted in the blocking buffer, and cells were incubated for 1 hour at room temperature. Additional information about immunofluorescence (IF) microscopy is included in supplemental Materials and methods.
Results
Identification of PfGARP as RBC-binding protein by phage display cDNA screens
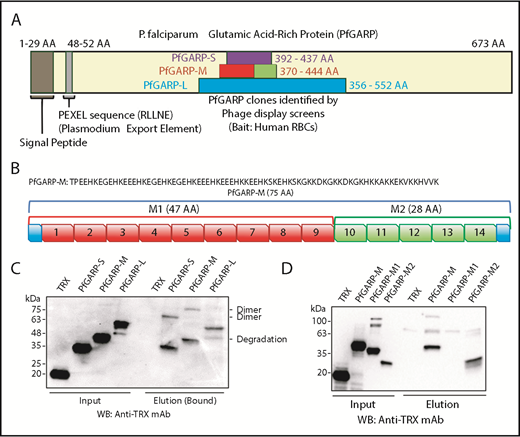
We have previously used the phage display cDNA technology to identify malaria parasite proteins that bind to the surface of intact RBCs.15,17,40 Here, we screened a phage display cDNA library of P falciparum 3D7 (sialic acid–independent strain) using human RBCs as bait. One of these clones isolated from multiple rounds of phage display screens was PfGARP (AAs 356-552), designated here as PfGARP-L (Figure 1A). We also performed an independent phage display screen using the sialic acid–dependent FCR3 strain and neuraminidase-treated RBCs. Interestingly, a smaller PfGARP clone (AAs 392-437), designated here as PfGARP-S (Figure 1A), was identified from multiple cDNA library screens. We considered independent isolation of overlapping PfGARP clones as a strong indication of its potential as an RBC-binding protein.
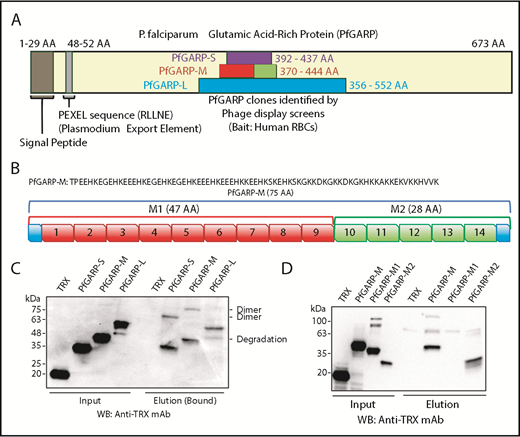
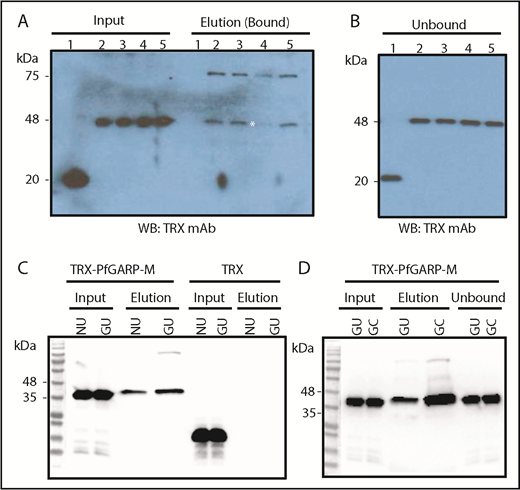
Identification of PfGARP by phage display screens and RBC-binding assay. (A) Schematic representation of PfGARP segments identified by phage display screens. PfGARP-L (blue) and PfGARP-S (purple) are 2 overlapping sequences obtained from independent phage display P falciparum screens using human RBCs as bait. PfGARP-M (red and green) is a codon-optimized stable segment. PfGARP-M1 (red) and PfGARP-M2 (green) are nonoverlapping segments of PfGARP-M protein. (B) A 75-AA acid sequence of PfGARP-M (3D7) was obtained from PlasmoDB site.76 PfGARP-M1 (red) consists of 9 repeats, and PfGARP-M2 (green) consists of 5 repeats. (C) Expression, purification, and RBC binding of TRX-PfGARP recombinant proteins. All PfGARP-S, PfGARP-M, and PfGARP-L proteins bound to RBCs. PfGARP-M showed the least degradation. (D) Expression, purification, and RBC binding of TRX–PfGARP-M1 and PfGARP-M2 proteins. PfGARP-M and PfGARP-M2 bound to RBCs but not PfGARP-M1. TRX served as a negative control. All protein concentrations were normalized at 1.0 μM, and NaCl (1.5 M) was used to elute RBC-bound proteins. Mouse monoclonal anti-TRX antibody was used to detect protein-binding signals.
Identification of PfGARP by phage display screens and RBC-binding assay. (A) Schematic representation of PfGARP segments identified by phage display screens. PfGARP-L (blue) and PfGARP-S (purple) are 2 overlapping sequences obtained from independent phage display P falciparum screens using human RBCs as bait. PfGARP-M (red and green) is a codon-optimized stable segment. PfGARP-M1 (red) and PfGARP-M2 (green) are nonoverlapping segments of PfGARP-M protein. (B) A 75-AA acid sequence of PfGARP-M (3D7) was obtained from PlasmoDB site.76 PfGARP-M1 (red) consists of 9 repeats, and PfGARP-M2 (green) consists of 5 repeats. (C) Expression, purification, and RBC binding of TRX-PfGARP recombinant proteins. All PfGARP-S, PfGARP-M, and PfGARP-L proteins bound to RBCs. PfGARP-M showed the least degradation. (D) Expression, purification, and RBC binding of TRX–PfGARP-M1 and PfGARP-M2 proteins. PfGARP-M and PfGARP-M2 bound to RBCs but not PfGARP-M1. TRX served as a negative control. All protein concentrations were normalized at 1.0 μM, and NaCl (1.5 M) was used to elute RBC-bound proteins. Mouse monoclonal anti-TRX antibody was used to detect protein-binding signals.
Based on a secondary structure–prediction algorithm, we designed a stable region of TRX–PfGARP-L, designated here as TRX–PfGARP-M (AA 370-444) (Figure 1A-B). An initial analysis of recombinant proteins indicated that TRX–PfGARP-M fusion protein was relatively more stable against proteolysis compared with TRX–PfGARP-L and TRX–PfGARP-S. Therefore, we used a codon-optimized PfGARP-M gene for recombinant expression in bacteria. The observed molecular weight (MW) of TRX–PfGARP-M without the TRX tag is ∼20 kDa, which is considerably higher than the calculated 9 kDa MW presumably due to the high percentage of charged residues and lack of defined secondary structure.41,42 Protein-binding measurements demonstrated that all 3 constructs of TRX-PfGARP (Figure 1C) can bind to intact RBCs (Figure 1C-D). In the RBC-sedimentation assays, higher MW bands were frequently detected, which most likely represent dimers or oligomers formed by the self-assembly of PfGARP under these conditions.
The TRX–PfGARP-M protein consists of a series of AA sequence repeats. We grouped these repeats into 2 regions: TRX–PfGARP-M1 (AAs 370-416) and TRX–PfGARP-M2 (AAs 417-444) (Figure 1B). TRX–PfGARP-M1 contains repeats 1 through 9 (shown in red), whereas TRX–PfGARP-M2 consists of repeats 10 through 14 (shown in green) (Figure 1B). Two nonoverlapping M1 and M2 regions were cloned from the codon-optimized TRX–PfGARP-M construct into the pET32 vector and expressed as TRX fusion proteins. Binding assays demonstrated that only TRX–PfGARP-M2 can directly bind to human RBCs, whereas no binding was detected with TRX–PfGARP-M1 or TRX under these conditions (Figure 1D). This mapping analysis narrowed down the core RBC-binding region of PfGARP to the M2 region.
Ectopic expression of PfGARP-M on CHO-K1 cells confers RBC recognition
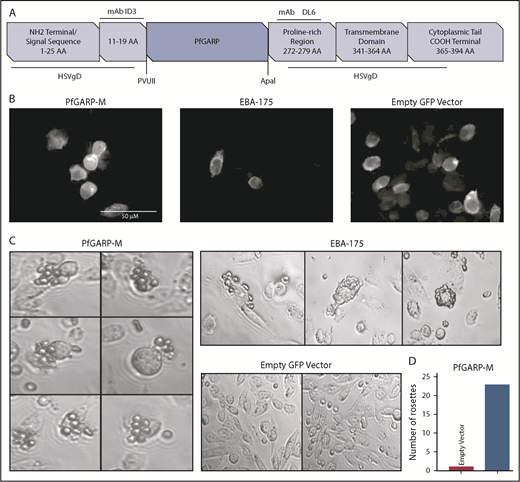
We expressed PfGARP-M on the surface of CHO-K1 cells (Figure 2A). The insertion site of PfGARP-M in pRE4 vector is flanked by mAb epitopes ID3 and DL6 (Figure 2A).28 The extracellular expression of the PfGARP insert on the surface of CHO-K1 cells was confirmed by IF using an anti-ID3 mAb (Figure 2B).28-30,43,44 EBA-175, a known microneme ligand that binds to RBCs,45 was transfected separately as a positive control. Multiple RBCs were recognized and captured by PfGARP-M–transfected CHO-K1 cells forming rosette-like structures (Figure 2C). The number of rosette-like structures was substantially greater in PfGARP-M–expressing cells than the empty pRE4 vector expressing GFP only (Figure 2C-D). Although the number of rosette-like structures formed by EBA-175–transfected CHO-K1 cells was relatively higher than the PfGARP-M–transfected cells, the general morphology of the structures was similar in both cases.30,46-48 These observations suggest that PfGARP-M expressed on the surface of heterologous cells can participate in adhesive interactions with uninfected RBCs and, perhaps, with nonerythroid cells, consistent with the formation of RBC aggregates induced by PfGARP peptides (supplemental Figure 1; Table 1).
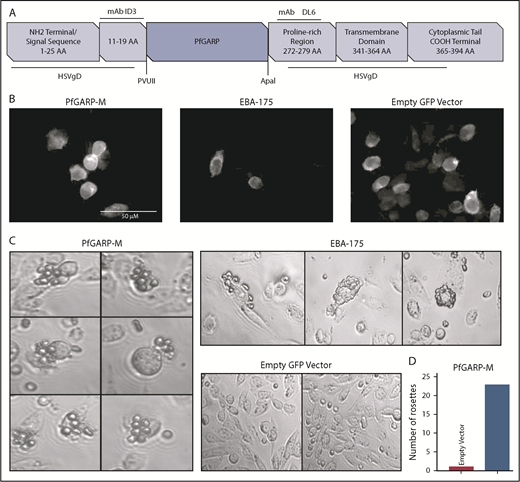
Expression of PfGARP on the surface of CHO-K1 cells. (A) Malaria proteins, PfGARP-M and EBA-175, were inserted into pRE4 vector, a chimeric construct of HSVgD1, and expressed on the surface of mammalian CHO-K1 cells. Location of epitopes for ID3 and DL6 are shown in the vector cartoon, which was adapted from Chitnis and Miller.28 (B) IF analysis of CHO-K1 cells transfected with PGARP-M, EBA-175, and empty vector confirming the surface expression of the respective proteins using mouse anti-ID3 mAb and goat anti-mouse immunoglobulin G antibody with Alexa Fluor 633. (C) CHO-K1 RBC-binding assay. Rosette-like structures were observed in PfGARP-M–transfected CHO-K1 cells (left panel; original magnification ×40). As expected, robust rosettes-like structures were observed in the EBA175-transfected CHO-K1 cells (upper right top panel), whereas no such structures were seen in CHO-K1 cells transfected with empty pRE4 vector (right lower panel). (D) A representative estimate of the rosette-like structures is shown in the bar graph. Quantification of rosette-like structures shown in panel C.
Expression of PfGARP on the surface of CHO-K1 cells. (A) Malaria proteins, PfGARP-M and EBA-175, were inserted into pRE4 vector, a chimeric construct of HSVgD1, and expressed on the surface of mammalian CHO-K1 cells. Location of epitopes for ID3 and DL6 are shown in the vector cartoon, which was adapted from Chitnis and Miller.28 (B) IF analysis of CHO-K1 cells transfected with PGARP-M, EBA-175, and empty vector confirming the surface expression of the respective proteins using mouse anti-ID3 mAb and goat anti-mouse immunoglobulin G antibody with Alexa Fluor 633. (C) CHO-K1 RBC-binding assay. Rosette-like structures were observed in PfGARP-M–transfected CHO-K1 cells (left panel; original magnification ×40). As expected, robust rosettes-like structures were observed in the EBA175-transfected CHO-K1 cells (upper right top panel), whereas no such structures were seen in CHO-K1 cells transfected with empty pRE4 vector (right lower panel). (D) A representative estimate of the rosette-like structures is shown in the bar graph. Quantification of rosette-like structures shown in panel C.
Immunodetection of native PfGARP in P falciparum–infected RBCs
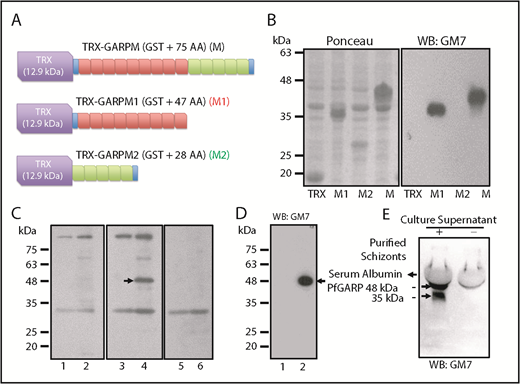
We generated an mAb (GM7) against PfGARP-M (see supplemental Materials and methods for details). To map the antigenic determinants, 2 nonoverlapping segments of PfGARP-M were expressed as TRX fusion proteins (Figure 3A-B). Western blotting detected TRX–PfGARP-M and TRX–PfGARP-M1 fusion proteins but not TRX–PfGARP-M2 protein (Figure 3B, right panel). We used mouse immune serum to detect native PfGARP in iRBC ghosts by western blotting (Figure 4C). Immune serum detected a 48-kDa band in iRBC ghosts but not in uninfected RBC ghosts (Figure 3C, arrow in lane 4). Serum reactivity of the 48-kDa band was completely blocked when immune serum was treated with His–PfGARP-M protein prior to western blotting (Figure 3C, lane 6). Other bands detected by the immune serum are likely to be nonspecific, because they were also detected by the preimmune serum (Figure 3C, lanes 1-2). Next, we used GM7 to detect native PfGARP in infected RBCs. GM7 detected a single 48-kDa band in iRBC ghosts (Figure 3D, arrow in lane 2). GM7 detected 2 bands corresponding to ∼48 kDa and ∼38 kDa in the culture supernatant (Figure 3E, arrows). The cellular localization of native PfGARP showed a punctate pattern in late trophozoite and schizont stages, and no signal was detected at the ring-stage of parasite culture (Figure 4). In some instances, a strong PfGARP signal was detected on the periphery of iRBCs. These results suggest that native PfGARP is detectable as clusters on the periphery of mature parasite-infected RBCs, as well as in the culture supernatant. Consistent with this model, a secretory form of PfGARP was detected in the supernatant of mature parasites during development (supplemental Figure 5).
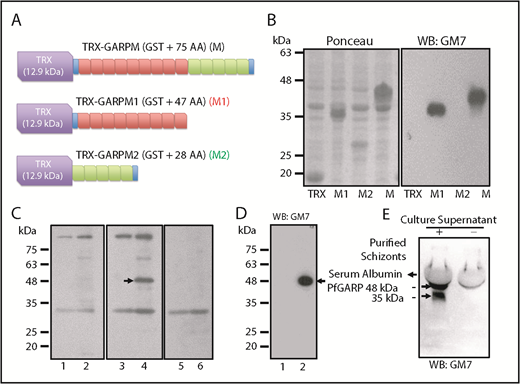
Characterization of GM7 mAb against PfGARP. (A) PfGARP-M was expressed as a fusion protein. TRX tag contributes ∼12.9 kDa, and PfGARP-M consists of 75 AAs. PfGARP-M1 consists of 9 repeats (47 AAs, red), and PfGARP-M2 consists of 5 repeats (28 AAs, green). Blue segments signify the flanking nonrepeat AAs (TPE and VVK) derived from PfGARP-M. (B) Ponceau staining of TRX, PfGARP-M1, PfGARP-M2, and PfGARP-M fusion proteins (left panel). Western blotting results of proteins shown in left panel (right panel). GM7 monoclonal specifically recognized PfGARP-M1 and PfGARP-M but not PfGARP-M2. (C) Detection of PfGARP in infected RBC ghosts. Normal RBC ghosts (lanes 1,3,5) and iRBC ghosts (lanes 2,4,6) were tested by western blotting using preimmune (lanes 1,2) and immunized (lanes 3-4) mouse serum against PfGARP-M. Immune serum detected an ∼48 kDa band (arrow, lane 3) but this band was not detected by preimmune serum (lane 2) in iRBC ghosts. Preincubation of immune serum with excess PfGARP-M prior to western blotting specifically blocked the detection of the 48 kDa band (lane 6). Preimmune serum detected several nonspecific bands. (D) Detection of 48-kDa PfGARP in iRBC ghosts. Normal RBC ghosts (lane 1) and iRBC ghosts (lane 2) were evaluated by western blotting using purified GM7 monoclonal. A single ∼48-kDa band was detected (lane 2, arrow). In fact, all hybridoma clones recognized the same band. Hybridoma clone #7 was selected and designated as GM7 for all subsequent studies. (E) Detection of native PfGARP in the P falciparum 3D7 culture supernatant of magnetically purified schizonts. Western blotting using GM7 detected 2 bands at ∼48 kDa and ∼38 kDa (arrows). The 38-kDa band represents a secondary processed secretory form of PfGARP or a degradation product of 48-kDa polypeptide detected in iRBC ghosts.
Characterization of GM7 mAb against PfGARP. (A) PfGARP-M was expressed as a fusion protein. TRX tag contributes ∼12.9 kDa, and PfGARP-M consists of 75 AAs. PfGARP-M1 consists of 9 repeats (47 AAs, red), and PfGARP-M2 consists of 5 repeats (28 AAs, green). Blue segments signify the flanking nonrepeat AAs (TPE and VVK) derived from PfGARP-M. (B) Ponceau staining of TRX, PfGARP-M1, PfGARP-M2, and PfGARP-M fusion proteins (left panel). Western blotting results of proteins shown in left panel (right panel). GM7 monoclonal specifically recognized PfGARP-M1 and PfGARP-M but not PfGARP-M2. (C) Detection of PfGARP in infected RBC ghosts. Normal RBC ghosts (lanes 1,3,5) and iRBC ghosts (lanes 2,4,6) were tested by western blotting using preimmune (lanes 1,2) and immunized (lanes 3-4) mouse serum against PfGARP-M. Immune serum detected an ∼48 kDa band (arrow, lane 3) but this band was not detected by preimmune serum (lane 2) in iRBC ghosts. Preincubation of immune serum with excess PfGARP-M prior to western blotting specifically blocked the detection of the 48 kDa band (lane 6). Preimmune serum detected several nonspecific bands. (D) Detection of 48-kDa PfGARP in iRBC ghosts. Normal RBC ghosts (lane 1) and iRBC ghosts (lane 2) were evaluated by western blotting using purified GM7 monoclonal. A single ∼48-kDa band was detected (lane 2, arrow). In fact, all hybridoma clones recognized the same band. Hybridoma clone #7 was selected and designated as GM7 for all subsequent studies. (E) Detection of native PfGARP in the P falciparum 3D7 culture supernatant of magnetically purified schizonts. Western blotting using GM7 detected 2 bands at ∼48 kDa and ∼38 kDa (arrows). The 38-kDa band represents a secondary processed secretory form of PfGARP or a degradation product of 48-kDa polypeptide detected in iRBC ghosts.
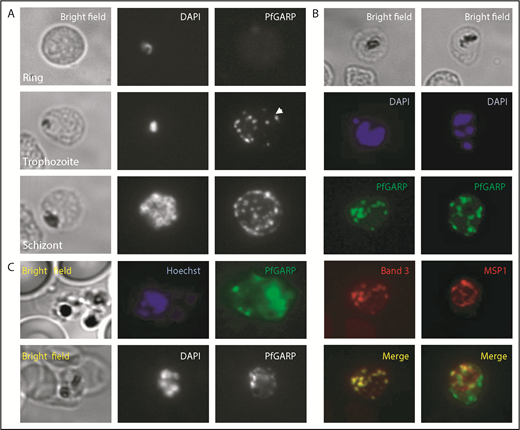
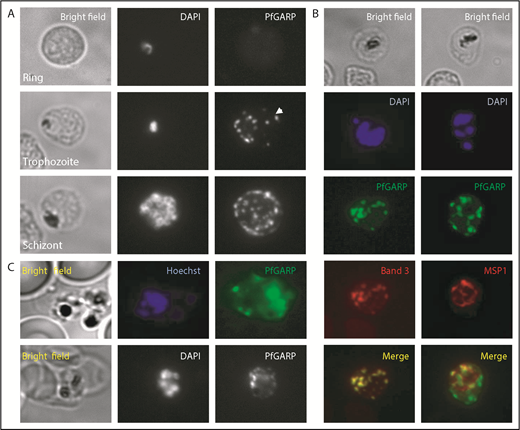
Localization of PfGARP in P falciparum–infected human erythrocytes. (A) PfGARP was detected in trophozoite and schizont stages of parasite infection but not in the ring stage. PfGARP appears as a punctate pattern particularly enriched at the erythrocyte periphery during the schizont stage of parasite development. White arrowhead shows exported PfGARP outside of the parasitophorous vacuole, consistent with the presence of a Plasmodium export element motif. (B) PfGARP colocalized with band 3 but not with MSP1, a merozoite surface protein. (C) Live parasite IF analysis suggesting that the PfGARP signal is detected on the surface of infected erythrocytes (upper panels). IF analysis under nonpermeabilizing conditions further suggests the surface localization of PfGARP (lower panels) (original magnification ×60).
Localization of PfGARP in P falciparum–infected human erythrocytes. (A) PfGARP was detected in trophozoite and schizont stages of parasite infection but not in the ring stage. PfGARP appears as a punctate pattern particularly enriched at the erythrocyte periphery during the schizont stage of parasite development. White arrowhead shows exported PfGARP outside of the parasitophorous vacuole, consistent with the presence of a Plasmodium export element motif. (B) PfGARP colocalized with band 3 but not with MSP1, a merozoite surface protein. (C) Live parasite IF analysis suggesting that the PfGARP signal is detected on the surface of infected erythrocytes (upper panels). IF analysis under nonpermeabilizing conditions further suggests the surface localization of PfGARP (lower panels) (original magnification ×60).
Consistent with our finding that erythrocyte band 3 is the host receptor for PfGARP, costaining for band 3 and PfGARP showed tight colocalization (Figure 4B, left panels). MSP1, a merozoite surface protein, was used as a negative control and showed no colocalization with PfGARP (Figure 4B, right panels). Our initial phage display screen used intact whole RBCs, suggesting PfGARP binding to the surface of RBCs. To demonstrate that the PfGARP signal seen in the IF studies of methanol-fixed parasitized cells is from the erythrocyte surface, we performed IF of live parasites (Figure 4C, upper panels) and nonpermeabilized fixed parasites (Figure 4C, lower panels). A punctate PfGARP pattern was observed in both cases, consistent with previous IF experiments.
PfGARP-M recognizes a chymotrypsin-sensitive receptor on human RBCs
To identify the host RBC receptor(s) recognized by PfGARP, human RBCs were treated with trypsin, chymotrypsin, and neuraminidase. Enzyme-treated RBCs were incubated with TRX or TRX–PfGARP-M, and the mixture was separated by centrifugation through silicone oil.49 Proteins bound to RBCs were eluted and analyzed by western blotting (Figure 5A-B). Binding of TRX–PfGARP-M to RBCs was reduced substantially only in chymotrypsin-treated human RBCs (Figure 5A). These results suggest that PfGARP binds to RBC receptors that are sensitive to chymotrypsin, and the PfGARP–RBC interaction is likely to be sialic acid independent. We measured the binding of TRX–PfGARP-M to GYPB-null human RBCs that lack the antigens carried by the GPB receptor (S-s-U-).50,51 TRX–PfGARP-M bound to normal and GYPB-null S-s-U- human RBCs (Figure 5C). We found a significant enhancement of TRX–PfGARP-M binding to GYPB-null S-s-U- RBCs that were treated with chymotrypsin compared with untreated GYPB-null S-s-U- RBCs (Figure 5D).
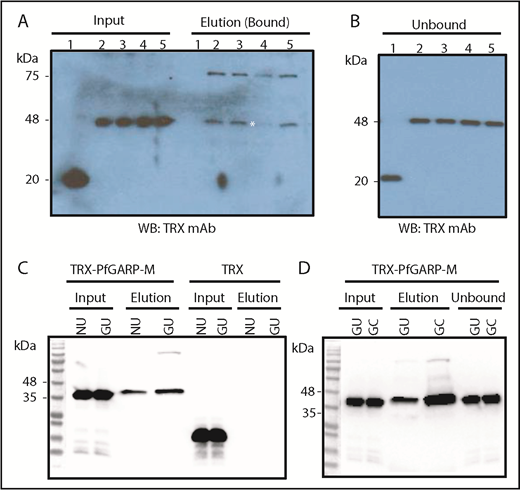
PfGARP binding to enzyme-treated RBCs and GYPB-null cells. (A) PfGARP-binding assay of enzyme-treated RBCs. TRX–PfGARP-M binding is sensitive to chymotrypsin treatment (elution lane 4; white asterisk). Input of proteins (left side) and bound proteins (right side) are shown. Lanes 1 and 2 show TRX and TRX–PfGARP-M in untreated human RBCs, respectively. Lanes 3 through 5 correspond to TRX–PfGARP-M in RBCs treated with neuraminidase (5.0 units/mL), chymotrypsin (1.0 mg/mL), and trypsin (1.0 mg/mL), respectively. (B) Unbound excess TRX and TRX–PfGARP-M are shown at equivalent amounts in all groups recovered from the binding assays. (C) Binding of TRX–PfGARP-M to normal and GYPB-null human RBCs. Normal human RBCs were used as positive control to demonstrate TRX–PfGARP-M binding. TRX–PfGARP-M binding to GYPB-null RBCs was similar to normal RBCs. In fact, a slight enhancement of TRX–PfGARP-M binding to GYPB-null RBCs was observed in several experiments. No binding was detected with the negative control (TRX). (D) Substantial enhancement of TRX–PfGARP-M binding toward chymotrypsin-treated GYPB-null human RBCs was detected. GC, GYPB-null chymotrypsin-treated human RBCs; GU, GYPB-null untreated human RBCs; NU, normal untreated.
PfGARP binding to enzyme-treated RBCs and GYPB-null cells. (A) PfGARP-binding assay of enzyme-treated RBCs. TRX–PfGARP-M binding is sensitive to chymotrypsin treatment (elution lane 4; white asterisk). Input of proteins (left side) and bound proteins (right side) are shown. Lanes 1 and 2 show TRX and TRX–PfGARP-M in untreated human RBCs, respectively. Lanes 3 through 5 correspond to TRX–PfGARP-M in RBCs treated with neuraminidase (5.0 units/mL), chymotrypsin (1.0 mg/mL), and trypsin (1.0 mg/mL), respectively. (B) Unbound excess TRX and TRX–PfGARP-M are shown at equivalent amounts in all groups recovered from the binding assays. (C) Binding of TRX–PfGARP-M to normal and GYPB-null human RBCs. Normal human RBCs were used as positive control to demonstrate TRX–PfGARP-M binding. TRX–PfGARP-M binding to GYPB-null RBCs was similar to normal RBCs. In fact, a slight enhancement of TRX–PfGARP-M binding to GYPB-null RBCs was observed in several experiments. No binding was detected with the negative control (TRX). (D) Substantial enhancement of TRX–PfGARP-M binding toward chymotrypsin-treated GYPB-null human RBCs was detected. GC, GYPB-null chymotrypsin-treated human RBCs; GU, GYPB-null untreated human RBCs; NU, normal untreated.
Identification of RBC receptor for PfGARP by blot overlay assay
The blot overlay assay has yielded successful results in the malaria field.34,52,53 Human RBC ghosts were evaluated by the blot overlay assay using PfGARP-M as a detector. Ghosts were made from normal untreated (NU) RBCs, normal chymotrypsin-treated RBCs, GYPB-null (S-s-U) untreated RBCs, and chymotrypsin-treated S-s-U- RBCs. The respective size of positive bands detected with TRX–PfGARP-M2 included an ∼48-kDa band in all 4 groups, an ∼60-kDa band in the chymotrypsin-treated ghosts from normal and GYPB-null human RBCs, and an ∼100 to 135–kDa band mainly in the GYPB-null RBC membranes (supplemental Figure 3, panel 4). The TRX–PfGARP-M protein showed comparable results (supplemental Figure 3, panel 3). No binding over the background was detected with the negative TRX control under the same conditions (supplemental Figure 3, panel 5).
The identification of a diffuse ∼100 to 135–kDa band in the GYPB-null RBC ghosts and detection of an ∼60-kDa band in the chymotrypsin-treated RBC ghosts gave a strong indication that band 3 is a potential host receptor for PfGARP. Chymotrypsin treatment of human RBCs is known to generate a stable ∼60-kDa fragment of band 3. Western blotting against the N-terminal cytoplasmic domain of band 3 detected the ∼100 to 135-kDa band and the ∼60-kDa band (supplemental Figure 3, asterisk in panel 7). The signal from the 48-kDa band was relatively weak, suggesting that this protein is a minor degradation product of band 3 or that PfGARP might recognize multiple receptors in human RBCs.
Band 3 is a host RBC receptor for PfGARP
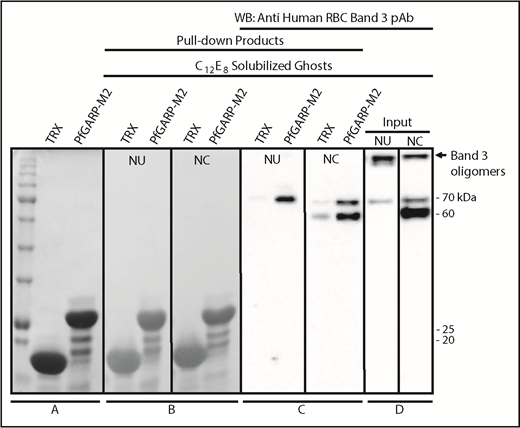
We performed pull-down assays to test whether band 3 can bind to PfGARP protein.35,36,54 TRX–PfGARP-M2 protein was immobilized on beads and incubated with C12E8 detergent–solubilized RBC ghosts. The C12E8 detergent was used to preserve the native conformation of band 3 and other membrane proteins. Western blotting confirmed that chymotryptic fragments of band 3 selectively bind to beads with immobilized PfGARP-M2 (Figure 6). Normal and chymotrypsin-treated solubilized ghosts were used in the pull-down assays. Some degradation of band 3 in the detergent-solubilized ghosts occurs as a result of the activation of RBC proteases.55-60 Similarly, formation of high-MW oligomers and release of an ∼70-kDa band are also known to occur in the C12E8-solubilized ghosts (Figure 6D).59 PfGARP-M2–immobilized beads also pulled down the 70-kDa fragment from untreated C12E8-solubilized RBC ghosts (Figure 6), whereas a prominent ∼60-kDa fragment, along with the 70-kDa fragment, was detected in the chymotrypsin-treated RBC ghosts (Figure 6D). The high-MW band 3 oligomers formed in the C12E8-solubilized ghosts were not captured by the PfGARP-M2–coated beads under these conditions. Together, these results demonstrate that PfGARP-M2 recognizes the chymotrypsin-sensitive ∼60-kDa segment of human erythrocyte band 3 as a host receptor.
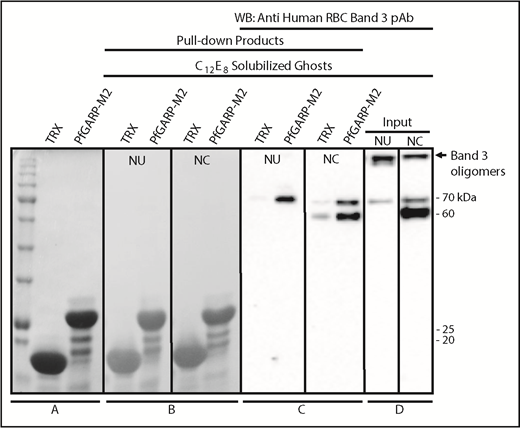
Identification of RBC band 3 binding to PfGARP using pull-down assay. TRX–PfGARP-M2 was used to pull down potential binding proteins from detergent-solubilized RBC ghosts. (A) Coomassie blue–stained 12% gel (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) showing TRX and TRX–PfGARP-M2 attached to cobalt beads. (B-C) TRX and TRX–PfGARP-M2 attached to beads were incubated with C12E8 solubilized normal untreated (NU) and chymotrypsin-treated (NC) ghosts. Bound proteins were transferred to nitrocellulose membrane. (Ponceau S staining). (C) Anti-band 3 western blotting. The 70-kDa band specifically associated with TRX–PfGARP-M2 in untreated RBCs (left panel). The 60-kDa and 70-kDa bands were detected in the chymotrypsin-treated RBCs (right panel). No binding of band 3 was detected with TRX-bound beads. (D) Western blot of C12E8-solubilized normal and chymotrypsin-treated ghosts as inputs. Western blotting of TRX–PfGARP-M2 and TRX proteins alone did not show any nonspecific signal detected by anti-band 3 antibody (data not shown).
Identification of RBC band 3 binding to PfGARP using pull-down assay. TRX–PfGARP-M2 was used to pull down potential binding proteins from detergent-solubilized RBC ghosts. (A) Coomassie blue–stained 12% gel (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) showing TRX and TRX–PfGARP-M2 attached to cobalt beads. (B-C) TRX and TRX–PfGARP-M2 attached to beads were incubated with C12E8 solubilized normal untreated (NU) and chymotrypsin-treated (NC) ghosts. Bound proteins were transferred to nitrocellulose membrane. (Ponceau S staining). (C) Anti-band 3 western blotting. The 70-kDa band specifically associated with TRX–PfGARP-M2 in untreated RBCs (left panel). The 60-kDa and 70-kDa bands were detected in the chymotrypsin-treated RBCs (right panel). No binding of band 3 was detected with TRX-bound beads. (D) Western blot of C12E8-solubilized normal and chymotrypsin-treated ghosts as inputs. Western blotting of TRX–PfGARP-M2 and TRX proteins alone did not show any nonspecific signal detected by anti-band 3 antibody (data not shown).
Quantification of PfGARP immune response in malaria-endemic areas
We developed a quantitative ELISA to detect antibodies against PfGARP. The GM7 mAb recognized the M1 repeats but not the M2 repeats (Figure 3A-B). To obtain the proof-of-principle evidence for the clinical relevance of the GM7 epitope present in PfGARP-M, and particularly within M1P6 peptide, we acquired 380 plasma samples from subjects living in rural areas of Kambila, Mali, where transmission of P falciparum is seasonal and intense.61,62 Plasma samples came from subjects representing both sexes, as well as young children, exposed to malaria. We first screened the plasma samples using purified His–PfGARP-M protein immobilized to an ELISA plate. This screen was designed to capture all positive hits targeting the epitopes located within PfGARP-M (supplemental Figure 4; Table 1). Next, we tested the same plasma samples for antibodies against PfGARP using immobilized M1P6 peptide. Consistent with the results from the His–PfGARP-M screen (supplemental Figure 4C), the M1P6-immobilized ELISA showed high antibody reactivity in the same plasma samples. In fact, background signal was substantially reduced in the M1P6-coated ELISA screen (supplemental Figure 4D). In summary, the peptide-based mapping of the reactive epitope recognized by GM7 mAb led to the development of M1P6 peptide–based ELISA, which is likely to allow quantification of the immune response against PfGARP in subjects exposed to malaria infection in endemic areas.
Further details can be found in “Additional results” in the supplemental Materials and methods.
Discussion
PfGARP was identified as a cDNA clone from the expression library of P falciparum isolate FC27 using sera from subjects living in Papua, New Guinea.19 Although the primary structure of the PfGARP gene predicted a mature protein of ∼77 kDa, the reactive sera from malaria patients failed to detect any endogenous protein for unknown reasons.19 A previous study identified PfGARP in a global gene-disruption strategy designed to find parasite proteins that are required for P falciparum erythrocyte membrane protein 1 trafficking and adhesion properties of iRBCs expressing the var2csa variant of P falciparum erythrocyte membrane protein 1 to chondroitin sulfate A.6 The disruption of the PfGARP gene had no effect on the viability or rigidity of iRBCs.6 Consistent with this observation, a more recent study used the piggyBac transposon insertional mutagenesis strategy and showed that disruption of the PfGARP gene does not affect parasite viability and survival.63 Using RNA sequencing of clinical isolates, Duffy and colleagues demonstrated a significant upregulation of PfGARP in parasites isolated from infected children but not pregnant women.23 Similarly, PfGARP is differentially regulated in adhesive P falciparum parasites, FCR-3–chondroitin sulfate A, and FCR-3–CD36.27,64 Because parasites isolated from infected children preferentially bind to CD36,65 these observations suggest that PfGARP mediates adhesion of iRBCs to multiple host receptors. Combined with the expression of PfGARP restricted to P falciparum, these findings imply that PfGARP plays a functional role in the adhesion properties that are unique to P falciparum–mediated pathogenesis. However, the molecular basis of PfGARP function in P falciparum adhesion pathways is not known.
In this article, we report the identification and mapping of an RBC binding site in PfGARP using multiple phage display screens from independent cDNA libraries of P falciparum. Multiple overlapping clones of PfGARP identified using normal RBCs and neuraminidase-treated RBCs indicate that PfGARP interacts with a sialic acid–independent site(s) on intact RBCs. PfGARP-M protein is composed of 2 distinct repeats containing glutamic acid or lysine-rich residues. Although RBC binding activity was not detected in PfGARP-M1, we cannot rule out the possibility that additional RBC binding sites exist outside the boundary of PfGARP-L. To further confirm the RBC-binding activity of PfGARP-M, we expressed PfGARP-M on CHO-K1 cells and demonstrated the formation of rosette-like structures with RBCs (Figure 2). Previously, this approach has been used to characterize the RBC-binding activity of malaria proteins.48 Finally, formation of RBC aggregates by peptides from PfGARP-M2 repeats, which contain the RBC-binding activity, suggests that PfGARP-M plays a functional role in promoting the adhesion properties of host RBCs.
Development of GM7 monoclonal and mapping of its epitope within PfGARP-M1 allowed us to design a quantitative M1P6 peptide–based ELISA for PfGARP. Although screening of 380 human plasma samples from a malaria-endemic region of Mali provides the proof-of-principle evidence for the feasibility of measuring a human immune response against PfGARP, no direct correlation between the functional role of PfGARP immune response and malaria protection can be inferred from this screen. Our anticipation is that a high antibody response against PfGARP may correlate with the protection against severe malaria, particularly symptoms of cerebral malaria in children with P falciparum malaria. If successful, these findings may identify PfGARP as a potential biomarker for the disease progression in severe malaria.
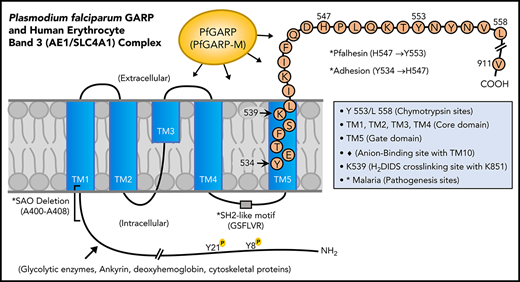
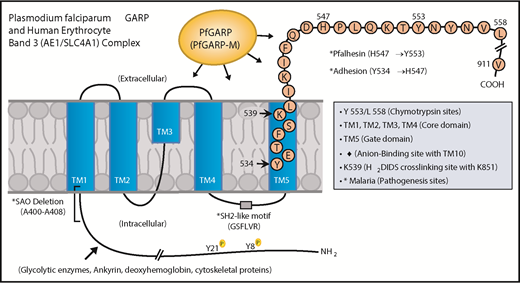
Domain mapping of PfGARP, including the identification of PfGARP-M2 as the critical segment that binds to the surface of intact RBCs, prompted us to search for its host receptor. It is known that chymotrypsin cleaves human RBC band 3 at 2 closely spaced sites (Y-553 and L-558) located in its third extracellular loop and leaves the multipass membrane protein functional and attached to the membrane66 (Figure 7). The pull-down assay suggests that band 3 is a host receptor for PfGARP and demonstrated that the ∼60-kDa fragment of band 3 contains the specific binding site for PfGARP. These results define the core RBC binding site of PfGARP and show that the N-terminal ∼60-kDa segment of band 3 serves as its cognate RBC receptor.
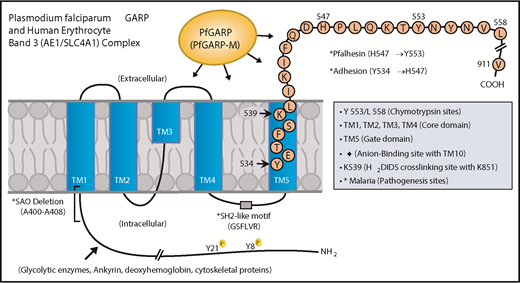
Proposed model of a functional role for PfGARP in malaria infection. PfGARP recognizes a site within the N-terminal 60-kDa ectoplasmic domain of human RBC band 3. This site is likely to be in close proximity to the region of band 3 that mediates RBC adhesion in malaria.67 Interestingly, several known features of malaria pathogenesis have been mapped to the same N-terminal domain of band 3. This cartoon was assembled from the recently published structure of human RBC band 3.77
Proposed model of a functional role for PfGARP in malaria infection. PfGARP recognizes a site within the N-terminal 60-kDa ectoplasmic domain of human RBC band 3. This site is likely to be in close proximity to the region of band 3 that mediates RBC adhesion in malaria.67 Interestingly, several known features of malaria pathogenesis have been mapped to the same N-terminal domain of band 3. This cartoon was assembled from the recently published structure of human RBC band 3.77
In a series of elegant studies, Sherman and other researchers have shown an intriguing role for modified band 3 as a malaria adhesion receptor expressed on the surface of iRBCs, suggesting clustering and oxidative environment as potential mediators.67-70 It was shown that modified band 3 promoted the adhesion of iRBCs specifically through its third ectodomain. Band 3 mAbs and synthetic peptides corresponding to AAs 537 to 547 and AAs 547 to 553 (termed Pfalhesin) and AAs 824 through 829 blocked adhesion of P falciparum iRBCs in vitro.67,71-73 Moreover, many late-stage P falciparum iRBCs were detected in the blood of Aotus and Saimiri monkeys 24 hours following the injection of band 3 synthetic peptides, indicating their adhesive/sequestration inhibitory effect in vivo.67,73,74 In fact, specific regions of modified band 3 were found to bind receptors such as CD36 and thrombospondin.73 Our findings demonstrate that PfGARP binds to band 3 in the same region as identified by Sherman and colleagues,67,68 thus raising the possibility that PfGARP may modulate binding of infected RBCs to pathogenic sites via CD36 and other receptors.75 Another possibility to consider is that PfGARP binds to band 3 and forms a stable sticky complex, which, in turn, interacts with other receptors on multiple cell types. This phenomenon could be further potentiated if PfGARP self-assembles and recruits additional band 3 receptors on normal RBCs and iRBCs. Such possibilities will now be experimentally testable in future studies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the following colleagues for their valuable contributions during the course of this study: Theresa Coetzer for sharing the phage display cDNA library used in the initial screens; Joshua Cole and Sheila Amoako for optimizing the phage display screens under the Tufts Building Diversity in Biomedical Sciences program; James Baleja for PSIPRED sequence analysis; Gary H. Cohen and Roselyn J. Eisenberg for providing mAbs ID3 and DL6; John Quigley, Nadim Mahmud, and Sally Campbell (University of Illinois at Chicago) and Jigna Rami (Red Cell Reference Laboratory, ITxM Clinical Services) for providing GYPB-null RBCs; Mark Diethelm (Tufts Medical Center) for providing RBCs from deidentified hemochromatosis patients; Mercio Perrin, David Greenblatt, James Baleja, and Carlo Brugnara for technical guidance to H.A.; Andrew Levin (Immunetics) for providing plasma from deidentified Babesia microti–infected patients and technical advice for ELISA; Allen Parmelee and Stephen Kwok (Tufts Core Facility) for help with flow cytometry; Jennifer Nwankwo for technical assistance with the CHO-K1–binding assay; David Liu and Cathy Korsgren for polyclonal antibody against the cytoplasmic N terminus of human RBC band 3; David Liu for many discussions during the course of this study; Peter Crompton (National Institutes of Health) for providing human sera from Mali; and T.H. and A.H.C. for generating PfGARP mAb GM7 with the technical assistance of Douglas Jefferson (Cell Essentials) for hybridoma cell fusion. The authors are particularly grateful to Donna-Marie Mironchuk for numerous contributions to the administrative organization of the project, proofreading, multiple improvements of figures, and drawing of the model cartoon.
This work was supported by King Abdulaziz University, Saudi Arabia, for graduate training of H.A., National Institutes of Health, National Heart, Lung, and Blood Institute grants HL060961, HL051445, and HL095050 (A.H.C.), and Tufts Collaborates Seed Grant Program (A.H.C.), as well as by partial support from the Nutrition-Global Development Program of the Bill & Melinda Gates Foundation to a research consortium led by Gerald Combs and Simin Meydani at USDA–Human Nutrition Research Center on Aging at Tufts University.
Authorship
Contribution: H.A. performed the phage display screens, cloning, protein purification and binding, malaria invasion, aggregation, rosetting, blot overlay, pull-down assays, PfGARP detection in parasite culture supernatant, and wrote the first draft of the manuscript; C.S. performed IF, PfGARP-secretion assays, and protein-purification experiments; T.H. and A.H.C. generated the GM7 mAb; T.H. constructed all phage display cDNA libraries and performed many cloning and expression studies; M.M.K. performed and optimized the immunoassay used to detect antibody reactivity against PfGARP in patients; D.C.G.M. provided the pRE4 control plasmids and technical guidance during the optimization of CHO-K1 cell–based assays; A.H.C. designed the peptides used to map the PfGARP binding sites and development of ELISA; J.S. cloned and purified recombinant His-tagged PfGARP and participated in the initial invasion assays; M.R.B. and Y.L. provided technical assistance with the RBC-binding assays; S.H. maintained malaria culture and purified schizonts; and A.H.C. organized the figures and wrote multiple drafts of the manuscript as the principal investigator.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Athar H. Chishti, Department of Developmental, Molecular, and Chemical Biology, Tufts University School of Medicine, 150 Harrison Ave, Room 714, Boston, MA 02111; e-mail athar.chishti@tufts.edu.