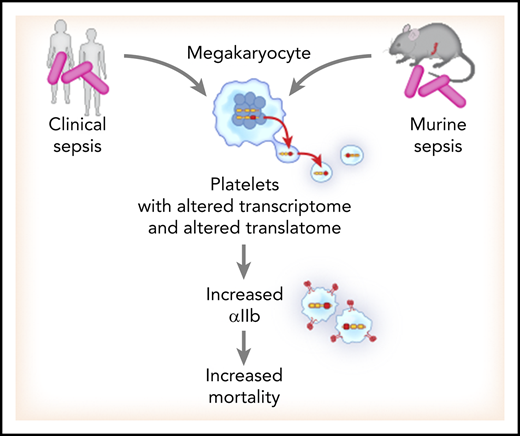
Key Points
Sepsis triggers transcriptional and translational alterations in platelets.
Sepsis-induced upregulation of ITGA2B predicts mortality.

Abstract
There is increasing recognition that platelets have a functional role in the pathophysiology of sepsis, though this role has not been precisely defined. Whether sepsis alters the human platelet transcriptome and translational landscape has never been established. We used parallel techniques of RNA sequencing and ribosome footprint profiling to interrogate the platelet transcriptome and translatome in septic patients and healthy donors. We identified 1806 significantly differentially expressed (false discovery rate <0.05) transcripts in platelets from septic patients. Platelet translational events during sepsis were also upregulated. To explore the relevance of a murine model of sepsis, cecal ligation and puncture (CLP), we compared sepsis-induced changes in platelet gene expression between septic patients and mice subjected to CLP. Platelet transcriptional (ρ = 0.42, P = 3.2 × 10−285) and translational (ρ = 0.65, P = 1.09 × 10−56) changes were significantly correlated between septic patients and mice. We focused on ITGA2B, tracking and validating the expression, regulation, and functional impact of changes in ITGA2B during sepsis. Increased ITGA2B was identified in bone marrow megakaryocytes within 24 hours of sepsis onset. Subsequent increases in ITGA2B were seen in circulating platelets, suggesting dynamic trafficking of the messenger RNA. Transcriptional changes in ITGA2B were accompanied by de novo protein synthesis of αIIb and integrin αIIbβ3 activation. Increased αIIb was associated with mortality in humans and mice. These findings provide previously unrecognized evidence that human and murine sepsis similarly alters the platelet transcriptional and translational landscape. Moreover, ITGA2B is upregulated and functional in sepsis due to trafficking from megakaryocytes and de novo synthesis in platelets and is associated with increased mortality.
Introduction
Sepsis affects ∼30 million individuals worldwide each year and remains one of the leading causes of infection-related hospitalizations and mortality.1 Sepsis may quickly progress to multiorgan failure and death.2,3 Alterations in platelet number and function are common in both clinical and experimental sepsis. Moreover, both thrombocytopenia and, independently, platelet hyperreactivity have been associated with increased sepsis-related morbidity and mortality.4,-6 Micro- and macrovascular thrombosis in sepsis further contributes to adverse outcomes.7 Sepsis survivors also have an increased long-term risk of thromboembolic events, including myocardial infarction and venous thromboembolism.8,-10
Although platelets are anucleate, an accumulating body of evidence demonstrates that the platelet transcriptome and proteome are not fixed. For example, platelets possess several pre-messenger RNAs (pre-mRNAs) and, in response to pathogens, bacterial products, and systemic agonists commonly generated during sepsis, splice these pre-mRNAs to generate mRNAs that are translated into proteins.11 More recent studies have further elucidated that the platelet transcriptome is altered in kidney disease, lupus, and myocardial infarction.12,-14
While the development of robust sequencing techniques, such as next-generation RNA sequencing (RNA-seq), has enhanced our ability to interrogate the human and mouse platelet transcriptome, unbiased global examination of translational events in platelets is more difficult to assess.15 RNA expression and protein abundance generally correspond with one another,15 and changes in RNA expression often predict changes in protein. However, protein synthesis is also regulated posttranscriptionally; therefore, RNA abundance only partly accounts for protein production. The advent of techniques employing deep sequencing of ribosome-protected mRNA fragments to assess active translation states has been a powerful tool employed in yeast and mammalian cells.16 Nevertheless, whether sepsis alters the human platelet transcriptional and translational landscape remains unknown. Additionally, whether commonly employed murine models of sepsis result in similar changes in platelet and megakaryocyte gene expression as observed in humans has never been established.
Here, we employed parallel techniques of RNA-seq and ribosome footprint profiling to interrogate the transcriptional and translational landscape in platelets from septic patients and murine platelets during experimental sepsis. We identified that numerous transcripts were altered in human and murine platelets during sepsis. Transcriptional changes were accompanied by increased de novo protein synthesis in platelets, as evidenced by ribosome footprint profiling and radiolabeling of newly translated proteins. Moreover, sepsis-induced transcriptional and translational changes in platelets were significantly conserved between humans and mice. Among the significantly altered transcripts in platelets during sepsis, ITGA2B (encoding αIIb protein) was one of the top upregulated transcripts, and our data suggest its mRNA is dynamically trafficked from bone marrow megakaryocytes to platelets >72 hours following sepsis induction. Further, during sepsis ITGA2B mRNA was actively translated to new protein in platelets, and this was accompanied by increased integrin αIIbβ3 activation. Increased ITGA2B in platelets during sepsis was associated with higher mortality in both humans and mice. Together, these data demonstrate that sepsis similarly alters the transcriptional and translational landscape of human and murine platelets, impacting functional responses and host outcomes.
Methods
Full details on methods can be found in supplemental Material (available on the Blood Web site).
Human subject enrollment
Patients admitted to 1 of 3 academic medical intensive care units (ICUs) with a primary diagnosis of severe sepsis or septic shock were prospectively enrolled within 48 (±24) hours of ICU admission. The institutional review boards approved this study. Each patient or a legally authorized representative provided written, informed consent. Sepsis was defined using the consensus criteria17 at the time this study was actively recruiting, defined as systemic infection and ≥2 of the following: (1) temperature >38°C or <36°C; (2) heart rate >90 beats/min; (3) respiratory rate >20 breaths/min or PaCO2 <32 mm Hg; and (4) white blood cell count >12 000 × 109/L, <4000 × 109/L, or >10% bands. Septic shock was defined as sepsis and the need for vasopressors. The prescription of antiplatelet agents (aspirin at any dose, clopidogrel, or nonsteroidal anti-inflammatory drugs) was recorded based on medication reconciliations done by medical staff upon ICU admission. For exclusion criteria, see supplemental Material. To examine the platelet transcriptome acutely and during recovery in a subset of septic patients, samples were collected from the same septic patients (n = 19) upon study enrollment (eg, acutely, after ICU admission for sepsis) and again in the same subjects ∼90 days after enrollment (eg, recovery from sepsis). In these longitudinal studies, age- and gender-matched, independently recruited, healthy donors (n = 10) were enrolled for comparison. Paired isolation of RNA and quantitative reverse transcription polymerase chain reaction (qRT-PCR) from septic patients and healthy donors occurred concurrently.
Next-generation RNA-seq
Platelets were isolated from septic patients and healthy donors within 30 minutes of blood sampling. The time from whole-blood collection to platelet isolation was similar between healthy donors and septic patients. Human and murine platelets were isolated and leukodepleted using our previously published methods that result in a highly purified population of <3 leukocytes/107 platelets (>99.9% purity) as counted by hemocytometer.18 Post-RNA-seq analysis of an index leukocyte transcript (PTPRC; CD45) confirmed that the samples were highly purified platelet preparations. For both human and mouse samples, we detected equivalent PTPRC levels between control and sepsis samples (P > .05) that were >5000 times lower in fragments per kilobase of transcript per million mapped reads (FPKM) than the index platelet transcript PF4. For next-generation RNA-seq, 1 × 109 isolated platelets were lysed in Trizol and then DNase treated. Total RNA was isolated and an Agilent bioanalyzer was used to quantify the amount and quality. RNA integrity number scores were similar between all samples (supplemental Table 5). RNA-seq libraries were prepared with TruSeq V2 with oligo-dT selection (Illumina, San Diego CA). Reads were aligned (Novoalign) to the reference genome at the time of these studies (GRCh37/hg19) and a pseudotranscriptome containing splice junctions. The Deseq2 analysis package was used to assign reads to composite transcripts (1 per gene) and quantitate FPKMs as previously described.18
Differential expression and significance were determined according to the Deseq2 algorithm, which controls for multiple comparisons using a false discovery rate (FDR) according to the method by Benjamini-Hochberg.19 Significant variance in expressed transcripts were prespecified as those transcripts with an FDR <0.05 and a log2 fold change ≥1.5 in sepsis, as compared with nonseptic conditions. For comparative analyses between humans and mice, only platelet transcripts with orthologs in both species and with expression >0.3 FPKM were considered. In these analyses, to allow for a broad cross-species comparison, we assessed all transcripts that were differentially expressed according to unadjusted P < .05. DAVID (https://david.ncifcrf.gov) was used for functional annotation analysis. RNA-seq data are publicly available (Bioproject PRJNA521077).
Ribosome footprint profiling
For ribosome footprint profiling experiments, 1 × 109 platelets were treated with cycloheximide (100 mg/mL) for 1 minute at room temperature to preserve ribosomes as natively attached to mRNAs. The platelets were lysed in Mammalian Lysis buffer (ARTseq, epicentre). Total RNA and ribosome-protected read RNAs (RPRs) were prepared per the manufacturer’s instructions (ARTseq-Ribosome Profiling Kit, epicentre). Sequencing libraries (25pM) were chemically denatured and applied to an Illumina HiSeq v4 single-read flow cell using an Illumina cBot. Hybridized molecules were clonally amplified and annealed to sequencing primers with reagents from an Illumina HiSeq SR Cluster Kit v4-cBot (GD-401-4001). Following transfer of the flow cell to an Illumina HiSeq 2500 instrument (HCSv2.2.38 and RTA v1.18.61), a 50-cycle single-read sequence run was performed using HiSeq SBS Kit v4 sequencing reagents (FC-401-4002). Ribosome profiling (eukaryote) library preparation was completed using the NEBNext Multiplex Small RNA Library Prep Set. Ribosome footprint profiling data are publicly available (Bioproject PRJNA521077).
Duolink proximity ligation assay technology assay for αIIb de novo protein synthesis
Antibodies against αIIb (target protein) and puromycin (which causes premature chain termination during translation) were cross-linked and visualized by confocal microscopy. Briefly, 1 × 108 human platelets were incubated with puromycin (in the presence or absence of anisomycin [negative control], translation inhibitor) for 20 minutes to capture αIIb de novo protein synthesis in situ. Platelets were fixed with 4% paraformaldehyde and spun down on coverglass. Platelets were permeabilized with 0.1% Triton, blocking serum was applied for 1 hour, and then platelets were incubated with the primary antibodies overnight (anti-CD41 [αIIb], Abcam, ab63983 and anti-puromycin, Kerafast, 3RH11). The manufacturer-recommended Duolink protocol (Sigma-Aldrich, DUO92007-30RXN) was used for signal amplification. Imaging was performed by confocal microscopy. Analysis was performed by CellProfiler imaging software and quantified for each imaged cell to avoid bias and exclude background staining.
Statistical analyses
Groups were compared using the Student t test or Wilcoxon rank sum (for continuous variables) and using the χ2 or Fisher’s exact test (for categorical variables), as appropriate (GraphPad Prism v7.0). More than 2 groups were compared using analysis of variance followed by Dunn’s multiple comparison test. A 2-tailed P value < .05 was considered statistically significant. For comparison of human and mouse transcripts, a Spearman correlation (ρ) was calculated.
Results
The platelet transcriptome is altered in human and experimental sepsis
To determine if sepsis alters the transcriptome of circulating human platelets, we performed RNA-seq on platelets from a discovery cohort of septic patients (n = 5) and matched healthy donors (n = 5). Principal-component analyses (PCAs) of total RNA expression identified differential grouping of septic patients compared with healthy donors along principal component 1, which accounted for 43% of the variance in transcript expression (Figure 1A). Differential expression analysis identified 1034 significantly upregulated and 772 significantly downregulated transcripts in platelets from septic patients compared with healthy donors (Figure 1B-C) at an FDR <0.05 and a log2 fold-change ≥1.5. These results suggest that there are marked alterations in the platelet transcriptome during sepsis.
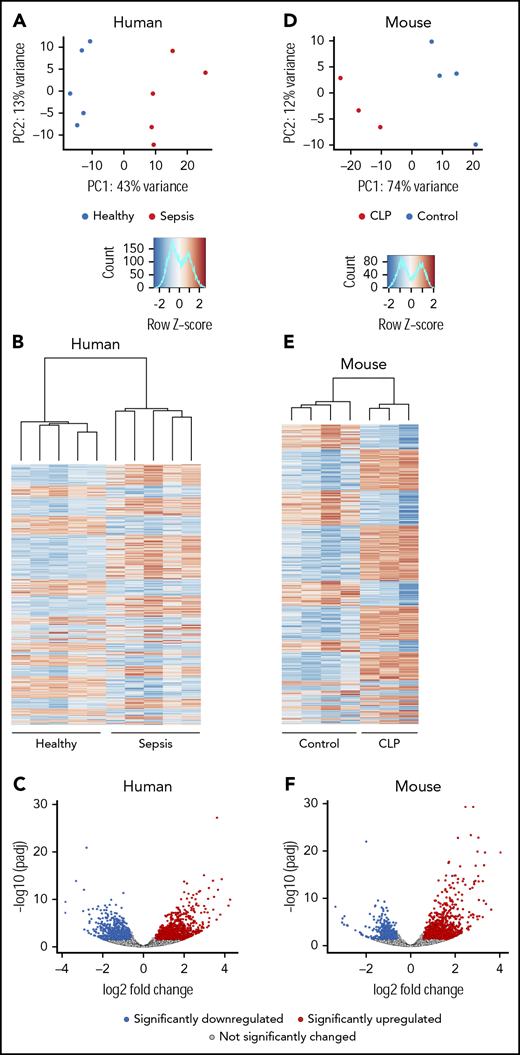
The human and murine platelet transcriptomes are altered in sepsis. (A-C) Platelets were isolated from septic patients within 48 hours of ICU admission or age-, race-, and gender-matched healthy donors. Total RNA was isolated, and next-generation RNA-seq was performed on platelets from septic patients and healthy donors, as described in “Methods” (n = 5/group). (A) PCA of the global platelet transcriptome in septic patients (red) and healthy donors (blue). (B) Heatmap and (C) volcano plot of significantly (FDR <0.05) upregulated (log2 fold-change >1.5, red) and downregulated (log2 fold-change <−1.5, blue) transcripts in platelets from septic patients. Black circles represent transcripts that were not significantly changed in sepsis. (D-F) Sepsis was induced in C57bl/J6 mice by CLP (n = 3, pooled platelets). For comparison, untreated C57bl/J6 mice were used as controls (n = 4). Platelets were isolated from CLP-treated mice on day 3 following surgery. Total RNA was isolated, and RNA-seq was performed on platelets from CLP and control mice, as described in “Methods.” (D) PCA of the global platelet transcriptome in CLP (red) and control mice (blue). (E-F) Heatmap and volcano plot of significantly (FDR <0.05) upregulated (log2 fold-change >1.5, red) and downregulated (log2 fold-change <−1.5, blue) transcripts in platelets from septic mice. Black circles represent transcripts that were not significantly changed in sepsis. padj, adjusted P value; PC, principal component.
The human and murine platelet transcriptomes are altered in sepsis. (A-C) Platelets were isolated from septic patients within 48 hours of ICU admission or age-, race-, and gender-matched healthy donors. Total RNA was isolated, and next-generation RNA-seq was performed on platelets from septic patients and healthy donors, as described in “Methods” (n = 5/group). (A) PCA of the global platelet transcriptome in septic patients (red) and healthy donors (blue). (B) Heatmap and (C) volcano plot of significantly (FDR <0.05) upregulated (log2 fold-change >1.5, red) and downregulated (log2 fold-change <−1.5, blue) transcripts in platelets from septic patients. Black circles represent transcripts that were not significantly changed in sepsis. (D-F) Sepsis was induced in C57bl/J6 mice by CLP (n = 3, pooled platelets). For comparison, untreated C57bl/J6 mice were used as controls (n = 4). Platelets were isolated from CLP-treated mice on day 3 following surgery. Total RNA was isolated, and RNA-seq was performed on platelets from CLP and control mice, as described in “Methods.” (D) PCA of the global platelet transcriptome in CLP (red) and control mice (blue). (E-F) Heatmap and volcano plot of significantly (FDR <0.05) upregulated (log2 fold-change >1.5, red) and downregulated (log2 fold-change <−1.5, blue) transcripts in platelets from septic mice. Black circles represent transcripts that were not significantly changed in sepsis. padj, adjusted P value; PC, principal component.
Next, we used cecal ligation and puncture (CLP) to establish whether coordinate changes occur in the murine platelet transcriptome under experimental sepsis conditions. RNA integrity number scores were similar among all conditions (septic patients, healthy donors, CLP-treated mice, and untreated mice) (supplemental Table 5). Similar to findings made in septic patients (Figures 1A-C), the experimental induction of sepsis triggered numerous changes in the murine platelet transcriptome. In PCA analysis, CLP samples grouped separately from controls across principal component 1, which accounted for 74% of the transcriptional variance. (Figures 1D-E). To identify significantly differentially expressed genes, and to maintain consistent filtering strategies between human and murine samples, we next restricted our analyses to only genes with an FDR <0.05 and a log2 fold-change ≥1.5 in expression. Through this strategy, we identified 890 significantly upregulated and 540 significantly downregulated transcripts in murine platelets during sepsis (Figure 1F). This is a similar number to findings in platelets from septic patients (n = 1806 transcripts; Figure 1B-C).
Sepsis-induced changes in the platelet transcriptome are similar in humans and mice
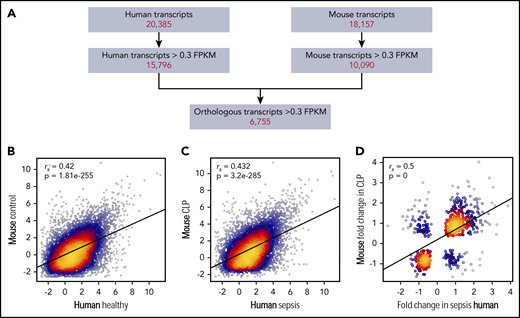
Whether murine sepsis results in similar platelet transcriptional changes as in human sepsis is unknown. Thus, we next combined our human and murine platelet transcriptional profiling data, retaining only those genes that were expressed (>0.3 FPKM20 ) in both human and murine platelets (Figure 2A). Through this strategy, we identified 6755 transcripts shared between human and murine platelets (Figure 2A); numbers similar to those previously reported by our group.20 Expression of these transcripts under nonseptic conditions (eg, healthy human donors and control mice) was significantly correlated (Figure 2B).

Alterations in the human platelet transcriptome during clinical sepsis are similar in murine platelets during experimental sepsis. (A) Filtering strategy for identifying orthologous and correlative human and mouse platelet transcriptome changes identified by RNA-seq. Of all transcripts identified by RNA-seq in human or murine platelets, we first restricted the analyses to transcripts with >0.3 FPKM. We next retained only those transcripts orthologous between the 2 species (n = 6755). (B) Correlation plot of the expression of conserved platelet transcripts in untreated control mice (n = 4) and healthy human donors (n = 5). (C) Correlation plot of the expression of conserved platelet transcripts in CLP-treated mice (n = 3, pooled platelets) and septic patients (n = 5). (D) Correlation plot of the change in expression (fold-change) of orthologous, significantly (P < .05) differentially expressed transcripts in both mouse and human platelets under septic conditions.
Alterations in the human platelet transcriptome during clinical sepsis are similar in murine platelets during experimental sepsis. (A) Filtering strategy for identifying orthologous and correlative human and mouse platelet transcriptome changes identified by RNA-seq. Of all transcripts identified by RNA-seq in human or murine platelets, we first restricted the analyses to transcripts with >0.3 FPKM. We next retained only those transcripts orthologous between the 2 species (n = 6755). (B) Correlation plot of the expression of conserved platelet transcripts in untreated control mice (n = 4) and healthy human donors (n = 5). (C) Correlation plot of the expression of conserved platelet transcripts in CLP-treated mice (n = 3, pooled platelets) and septic patients (n = 5). (D) Correlation plot of the change in expression (fold-change) of orthologous, significantly (P < .05) differentially expressed transcripts in both mouse and human platelets under septic conditions.
We then asked whether changes in the platelet transcriptome during sepsis were similar between humans and mice. Using the filtering strategy described above (Figure 2A), we identified a total of 3131 transcripts in either human or murine platelets that were significantly differentially expressed during sepsis. Sepsis-induced alterations in the platelet transcriptome were significantly correlated between humans and mice (Figure 2C). Of the transcripts differentially expressed in human and murine platelets during sepsis, 542 were shared between species and also concordantly changed (ie, upregulated or downregulated) in both humans and mice. Changes in these 542 differentially expressed transcripts in platelets during sepsis were significantly correlated between humans and mice (Figure 2D). Functional annotation analysis revealed upregulation in transcripts involved in membrane, cell-cell adhesion junction, focal adhesion, and cytoplasm activities. These changes indicate upregulation of genes involved in typical platelet functional responses and that may also be involved in more noncanonical roles under septic conditions.21 Additionally, these data illustrate a number of significantly differentially expressed genes that have recognized roles in the pathobiology of sepsis but have not been specifically investigated in platelets during sepsis. Some of these include CALR (encoding calreticulin22 ), PIM1 (encoding Pim-1, an antiapoptotic protein23 ), and HPSE (encoding heparanase24,25 ). A full list can be found in supplemental Table 2.
Platelet translational events are coordinately upregulated in clinical and experimental sepsis
Platelets participate in mRNA translation and de novo protein synthesis, often in regulated signal-dependent mechanisms.11,26,-28 Our data indicate that the transcriptional landscape of human and murine platelets is markedly altered in sepsis, with numerous genes undergoing significant changes in their expression (Figures 1,2-3). Our prior work identified that sepsis, as well as agonists generated during sepsis, induce the translation of several pre-mRNAs endogenously resident in human platelets.11,27 However, whether sepsis triggers global changes in the translational landscape of platelets and whether these changes are similar in humans and mice has never been examined.
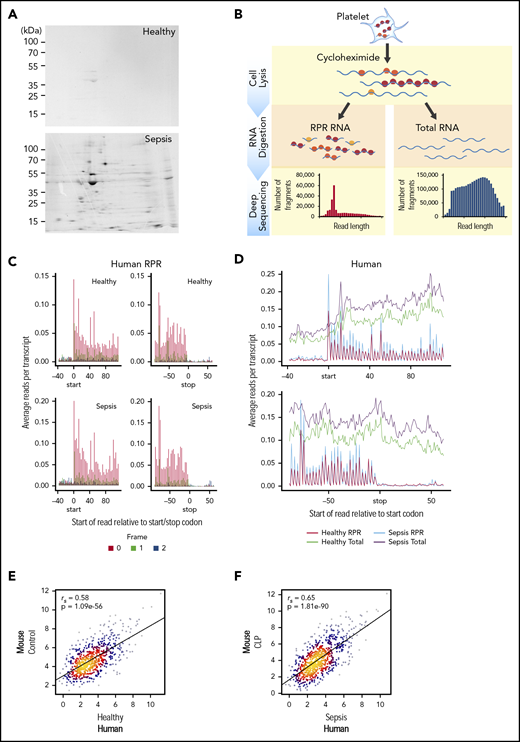
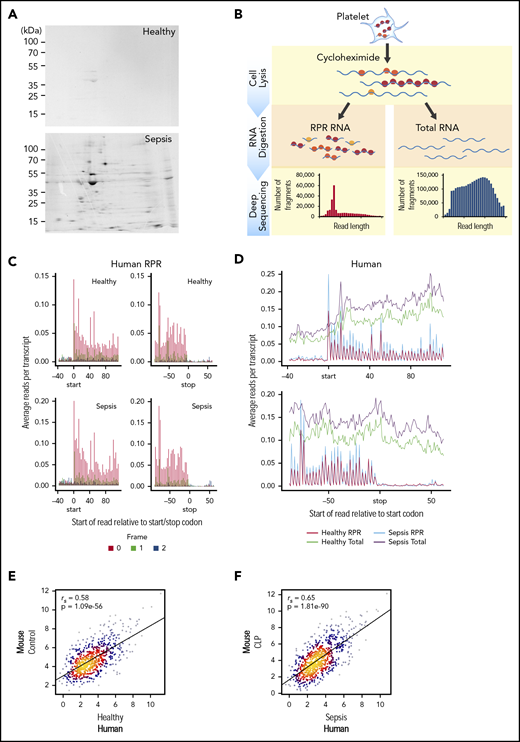
Translational events in platelets are increased coordinately in clinical and experimental sepsis. (A) Isolated platelets from septic patients or matched, healthy donors were incubated with 35S-methionine (t = 18 hours) to label newly synthesized proteins and then analyzed by 2D gel electrophoresis. Representative 35S-methionine–labeled 2D gel of platelets from a septic patient and healthy donor (representative of n = 3/group). (B) Schematic of protocol for sequencing total RNA (RNA-seq) and ribosome protected mRNA reads (RPRs, ribosome footprint profiling) in isolated human and murine platelets. (C) Ribosome footprint profiling of platelets from healthy donors and septic patients (n = 3/group) demonstrating global triplet periodicity. Increased reads align with frame 0 (red) compared with frame 1 (green) and frame 2 (blue). Frame number indicates distance from the first nucleotide of the P site. (D) Depiction of global number of reads per transcript for total RNA (healthy, green; sepsis, purple) and RPR (healthy, red; sepsis, blue) relative to the start and stop codon. (E) Correlation plot of the expression of orthologous, conserved RPRs in platelets from untreated control mice (n = 4) and healthy human donors (n = 3). (F) Correlation plot of the expression of orthologous, conserved RPRs in platelets from CLP-treated mice (n = 3, pooled platelets) and septic patients (n = 3).
Translational events in platelets are increased coordinately in clinical and experimental sepsis. (A) Isolated platelets from septic patients or matched, healthy donors were incubated with 35S-methionine (t = 18 hours) to label newly synthesized proteins and then analyzed by 2D gel electrophoresis. Representative 35S-methionine–labeled 2D gel of platelets from a septic patient and healthy donor (representative of n = 3/group). (B) Schematic of protocol for sequencing total RNA (RNA-seq) and ribosome protected mRNA reads (RPRs, ribosome footprint profiling) in isolated human and murine platelets. (C) Ribosome footprint profiling of platelets from healthy donors and septic patients (n = 3/group) demonstrating global triplet periodicity. Increased reads align with frame 0 (red) compared with frame 1 (green) and frame 2 (blue). Frame number indicates distance from the first nucleotide of the P site. (D) Depiction of global number of reads per transcript for total RNA (healthy, green; sepsis, purple) and RPR (healthy, red; sepsis, blue) relative to the start and stop codon. (E) Correlation plot of the expression of orthologous, conserved RPRs in platelets from untreated control mice (n = 4) and healthy human donors (n = 3). (F) Correlation plot of the expression of orthologous, conserved RPRs in platelets from CLP-treated mice (n = 3, pooled platelets) and septic patients (n = 3).
To answer this question, we used 35S-methionine radiolabeling followed by 2-dimensional (2D) electrophoresis to examine de novo protein synthesis in human platelets during sepsis. As shown in Figure 3A, global protein synthesis was increased in platelets isolated from septic patients compared with healthy donors. Samples were normalized to platelet count and total protein detected by Coomassie brilliant blue (supplemental Figure 1). We next used ribosome footprint profiling of platelets from septic patients and matched healthy donors to quantify translational events in platelets. A schematic of this approach is shown in Figure 3B.
In platelets from both septic patients and healthy donors, we identified numerous transcripts (n = 2950) with ≥1 RPR, consistent with ongoing translation of nascent mRNAs. These RPRs were of the expected size for mammalian cells, accumulated at the first nucleotide of the start codon, ended at the stop codon, and displayed characteristic triplet periodicity (Figure 3C and not shown). These features indicate that our ribosome footprint profiling approach was successful. Consistent with our observations by 35S-methionine labeling (Figure 3A), the number of RPRs was also increased in platelets from septic patients compared with healthy donors, indicating global upregulation of translational events during sepsis (Figure 3D). Similar global increases in translation were also seen in murine platelets following CLP (supplemental Figure 2A-B).
The expression of RPRs in platelets was significantly correlated between healthy human donors and control mice (Figure 3E). Interestingly, the strength of the association between human and murine platelets was greater for RPRs (ρ = 0.58, P = 1.08e−56) Figure 3E) than for total RNA levels (ρ=0.42, P = 1.81e−255; Figure 2B). We next examined whether RPRs were also correlated in human and murine platelets during sepsis. Indeed, as shown in Figure 3F, the expression of RPRs in platelets was also significantly correlated between septic patients and mice following CLP. Moreover, the strength of this association in translational events (ρ = 0.65, P = 1.81e−90) was higher than for transcriptional events (ρ = 0.50, P = 0 Figure 2). These data suggest that consistent with sepsis-induced transcriptional changes in human and murine platelets (Figure 2), the translational landscape of human and murine platelets is similarly altered.
Integrin subunit αIIb (ITGA2B) expression is upregulated in platelets during sepsis
αIIbβ3 is a platelet- and megakaryocyte-specific integrin regulating, among other things, adhesion to inflamed endothelial cells and prothrombotic responses.29,30 We observed that one of the top upregulated transcripts in platelets from septic patients, compared with healthy donors, was ITGA2B, which encodes for the αIIb subunit of integrin αIIbβ3 (Figure 4A; supplemental Table 2). Similarly, sequencing of ribosome protected mRNAs in platelets from septic patients demonstrated that ITGA2B was one of the highest enriched transcripts with ribosome protected fragments (supplemental Table 2) and that translation of ITGA2B was significantly increased in septic patients (supplemental Figure 3A). Expression of ITGB3 (which encodes for the β3 subunit) was also significantly upregulated in platelets during sepsis (supplemental Figure 3B-C), although this transcript is expressed by other cells in addition to platelets. We next focused on validating the transcriptional and translational changes, and functional significance of αIIb protein in platelets during sepsis. To confirm the reproducibility of our RNA-seq findings, we measured the expression of ITGA2B by qRT-PCR in a larger cohort of septic patients (n = 94) and healthy donors (n = 37). The characteristics and clinical data of septic individuals and healthy donors are shown in Table 1 and supplemental Table 3. Approximately 55% of septic patients also had shock, and 62% required mechanical ventilation, consistent with a high severity of illness. As expected, platelet counts in septic patients were significantly lower compared with healthy donors.

ITGA2B is upregulated in human and murine platelets during clinical and experimental sepsis. (A) Integrated Genomics Viewer (IGV) image showing the expression of ITGA2B exonal reads in purified platelets from a healthy donor (top) vs a septic patient (bottom). The thicker bars on the bottom represent coding regions of ITGA2B with 5′ and 3′ ends indicated. The y-axis represents the expression of ITGA2B, with higher peaks indicating increased mRNA expression. Images representative of n = 5 subjects per group. (B) qRT-PCR of ITGA2B expression in platelets from septic patients (n = 94) assessed upon ICU admission (study day 1) and matched healthy donors (n = 37). Data show the fold change of ITGA2B expression in septic patients vs healthy donors. (C) Serial qRT-PCR measurements of ITGA2B in platelets from an independent cohort of septic patients assessed upon admission to the ICU (day 1, septic n = 19) and again in the same patients ∼90 days following hospital discharge (day 90, n = 19). For comparison, ITGA2B expression was also measured in an independent cohort of matched healthy donors (Healthy, n = 10). (D) IGV image showing Itga2b exonal reads in platelets from untreated control mice (top) and mice subject to CLP (bottom). The thicker bars on the bottom represent coding regions of Itga2b with 5′ and 3′ ends indicated. The y-axis represents the expression of Itga2b, with higher peaks indicating increased RNA expression. Images are representative of CLP (n = 3 biological replicates) and control (n = 4 biological replicates). (E) Serial measurements by qRT-PCR of Itga2b in primary bone marrow–derived megakaryocytes from control or CLP mice on days 1 and 3 (n = 4-13/ group). (F) Serial measurements by qRT-PCR of Itga2b in isolated platelets from untreated (control) or post-CLP mice on days 1, 2, 3, and 7. Day 7 post-CLP was considered a recovery time point (n = 6-10/group).
ITGA2B is upregulated in human and murine platelets during clinical and experimental sepsis. (A) Integrated Genomics Viewer (IGV) image showing the expression of ITGA2B exonal reads in purified platelets from a healthy donor (top) vs a septic patient (bottom). The thicker bars on the bottom represent coding regions of ITGA2B with 5′ and 3′ ends indicated. The y-axis represents the expression of ITGA2B, with higher peaks indicating increased mRNA expression. Images representative of n = 5 subjects per group. (B) qRT-PCR of ITGA2B expression in platelets from septic patients (n = 94) assessed upon ICU admission (study day 1) and matched healthy donors (n = 37). Data show the fold change of ITGA2B expression in septic patients vs healthy donors. (C) Serial qRT-PCR measurements of ITGA2B in platelets from an independent cohort of septic patients assessed upon admission to the ICU (day 1, septic n = 19) and again in the same patients ∼90 days following hospital discharge (day 90, n = 19). For comparison, ITGA2B expression was also measured in an independent cohort of matched healthy donors (Healthy, n = 10). (D) IGV image showing Itga2b exonal reads in platelets from untreated control mice (top) and mice subject to CLP (bottom). The thicker bars on the bottom represent coding regions of Itga2b with 5′ and 3′ ends indicated. The y-axis represents the expression of Itga2b, with higher peaks indicating increased RNA expression. Images are representative of CLP (n = 3 biological replicates) and control (n = 4 biological replicates). (E) Serial measurements by qRT-PCR of Itga2b in primary bone marrow–derived megakaryocytes from control or CLP mice on days 1 and 3 (n = 4-13/ group). (F) Serial measurements by qRT-PCR of Itga2b in isolated platelets from untreated (control) or post-CLP mice on days 1, 2, 3, and 7. Day 7 post-CLP was considered a recovery time point (n = 6-10/group).
As shown in Figure 4B, there was an approximately ninefold increase in platelet ITGA2B expression in septic patients compared with healthy donors (P < .0001). This was accompanied by increased basal integrin αIIbβ3 activation on platelets from septic patients (supplemental Figure 4A-B). To determine whether increases in ITGA2B expression during clinical sepsis are sustained or resolve over time, we prospectively followed sepsis patients during their ICU stay and, in surviving sepsis patients, examined ITGA2B expression ∼90 days following hospital discharge. In septic patients, ITGA2B expression peaked at ICU day 7 but remained significantly elevated relative to matched healthy donors (supplemental Figure 4E). In a subset of septic patients (n = 19), ITGA2B expression 90 days after enrollment was comparable to healthy donors (Figure 4C). This indicates that sepsis upregulates ITGA2B acutely and that these gene expression changes resolve upon recovery from sepsis in humans.
Increased ITGA2B during sepsis is invested into platelets by bone marrow megakaryocytes
We next sought to identify the mechanisms by which platelet ITGA2B expression increases in platelets during sepsis. We hypothesized that initially, sepsis triggers megakaryocytes to invest greater ITGA2B mRNA content into developing platelets. To test this, we performed CLP in mice and measured ITGA2B expression in platelets and autologous, primary, bone marrow megakaryocytes isolated from the same animals on sequential days following CLP. ITGA2B expression increased in platelets following induction of murine sepsis via CLP (Figure 4D). We observed significant and early increases in megakaryocyte ITGA2B expression 24 hours post-CLP (day 1), followed 72 hours later by upregulation in platelet ITGA2B expression (day 3, Figure 4E-F). These findings suggest that upregulation of ITGA2B in platelets during sepsis is a result of increased ITGA2B transcription in megakaryocytes, leading to increased mRNA content in developing and newly released platelets. Similar to observations in patients who had recovered from sepsis (Figure 4C), platelet ITGA2B expression in mice normalized 7 days following CLP - sufficient time to have near-complete murine platelet turnover (Figure 4F).31
Integrin subunit αIIb is newly synthesized in platelets during sepsis
Our findings indicate that the transcriptional and translational landscapes in platelets are altered during sepsis (Figures 1,2-3), and that megakaryocytes invest platelets with increased ITGA2B mRNA content during sepsis (Figure 4). Thus, we next determined whether translation of ITGA2B mRNA, resulting in increased de novo synthesis of integrin αIIb protein, was upregulated in platelets during sepsis. Ribosome occupancy of ITGA2B mRNA was enriched in septic patients, with the ribosome-protected fragments exhibiting a clear trinucleotide periodicity (Figure 5A-B; supplemental Table 2 and supplemental Figure 3A). This suggested that active translation of ITGA2B mRNA in platelets was increased during human sepsis.
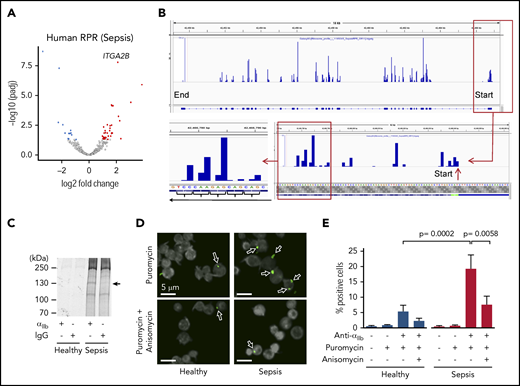
De novo translation of αIIb protein is increased in human platelets during clinical sepsis. Platelets were isolated from septic patients or matched healthy donors. (A) Volcano plot showing significantly, differentially expressed (FDR <0.05) ribosomal protected mRNA reads (RPRs) in platelets from septic patients or healthy donors (n = 5/group). ITGA2B is labeled. (B) Magnification of IGV file demonstrating triplet periodicity for ITGA2B. Here, ribosomes read the mRNA in groups of 3 nucleotides (a triplet) at a time where each triplet codon represents an amino acid code. mRNAs undergoing active translation will have ribosome reads generally stacked on the first nucleotide of each codon across the mRNA. This is termed triplet periodicity. (C) Newly synthesized αIIb protein (arrow) in platelets from septic patients detected by radiolabel (35S-methionine) and immunoprecipitation using an anti-αIIb antibody. An immunoglobulin G isotype-matched antibody was used as a control (representative of n = 3/group). (D) Confocal microscopy image of Duolink proximity ligation assay with puromycin demonstrating de novo translation of αIIb protein (green, arrows) in platelets from septic patients and healthy donors (n = 3/group). Anisomycin inhibits protein synthesis and was used here to demonstrate specificity of this system (eg, reduced staining for αIIb in the presence of anisomycin). (E) Quantification of percent positive cells for αIIb protein by CellProfiler, as described in “Methods.”
De novo translation of αIIb protein is increased in human platelets during clinical sepsis. Platelets were isolated from septic patients or matched healthy donors. (A) Volcano plot showing significantly, differentially expressed (FDR <0.05) ribosomal protected mRNA reads (RPRs) in platelets from septic patients or healthy donors (n = 5/group). ITGA2B is labeled. (B) Magnification of IGV file demonstrating triplet periodicity for ITGA2B. Here, ribosomes read the mRNA in groups of 3 nucleotides (a triplet) at a time where each triplet codon represents an amino acid code. mRNAs undergoing active translation will have ribosome reads generally stacked on the first nucleotide of each codon across the mRNA. This is termed triplet periodicity. (C) Newly synthesized αIIb protein (arrow) in platelets from septic patients detected by radiolabel (35S-methionine) and immunoprecipitation using an anti-αIIb antibody. An immunoglobulin G isotype-matched antibody was used as a control (representative of n = 3/group). (D) Confocal microscopy image of Duolink proximity ligation assay with puromycin demonstrating de novo translation of αIIb protein (green, arrows) in platelets from septic patients and healthy donors (n = 3/group). Anisomycin inhibits protein synthesis and was used here to demonstrate specificity of this system (eg, reduced staining for αIIb in the presence of anisomycin). (E) Quantification of percent positive cells for αIIb protein by CellProfiler, as described in “Methods.”
Consistent with this, the de novo synthesis of αIIb protein, as assessed by an 35S-radiolabeled immune-precipitation assay, was increased in platelets from septic patients compared with healthy donors (Figure 5C). This was further confirmed utilizing Duolink technology32 to fluorescently label newly translated αIIb protein in human platelets. As shown in Figure 5D-E, the number of platelets with newly translated αIIb protein was significantly increased in septic patients compared with healthy donors. In mice subject to CLP, we similarly found increased ribosome occupancy of Itga2b mRNA (supplemental Figure 5A-B), suggesting that in mice, as in humans, sepsis upregulates the synthesis of αIIb protein in platelets.
As targeting integrin αIIbβ3 has improved outcomes in some sepsis studies,29,30 we next examined associations between increased ITGA2B expression in platelets and adverse outcomes in sepsis. Consistent with prior reports,33 mortality was more than threefold higher in septic patients with thrombocytopenia upon ICU admission (Figure 6A). Thrombocytopenia was also modestly but significantly and inversely associated with platelet size (eg, mean platelet volume [MPV]), suggesting a release of new platelets during sepsis (Figure 6B). These findings in septic patients were not due to differences in patient age or illness severity (not shown). Consistent with the release of new platelets, MPV was significantly and positively associated with the expression of ITGA2B (Figure 6C). Increase ITGA2B expression in platelets from septic patients could not be attributed solely to increased platelet size in sepsis, as the MPV in septic patients and healthy donors was similar (supplemental Figure 6). Levels of ITGA2B expression were almost twofold higher in patients who did not survive sepsis, although differences did not reach statistical significance (Figure 6D). We next examined αIIb protein expression experimentally in mice subject to CLP. Whole blood was sampled from the tail vein and flow cytometry was performed following CLP to measure platelet surface expression of αIIb, and mice were monitored for survival over 96 hours (Figure 6E). Consistent with our findings of ITGA2B transcript levels in platelets from septic patients (Figure 6D), the expression of αIIb protein on platelets following CLP was significantly higher in animals that did not survive (Figure 6F).
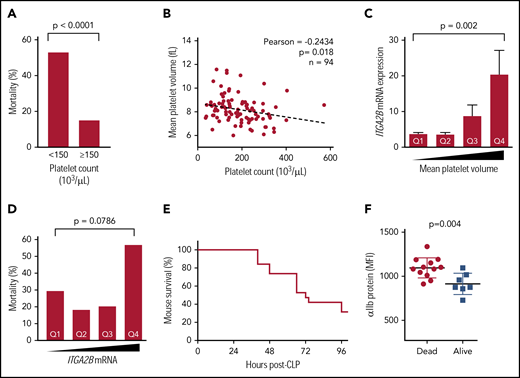
Increased ITGA2B and αIIb expression in sepsis is associated with both clinical and experimentally induced sepsis-related mortality. (A) In septic patients (n = 94), sepsis-related mortality is more than threefold higher in patients with thrombocytopenia at ICU admission (day 1). (B) Platelet counts in septic patients are inversely associated with MPV. (C) ITGA2B expression is significantly higher in newly released platelets (ie, platelets with a larger MPV) from septic patients. Quartiles for MPV were determined a priori and assigned as follows: quartile 1 (Q1), MPV <7.5 fL; Q2, MPV between 7.6 and 8.1 fL; Q3, MPV between 8.2 and 8.7 fL; Q4, MPV >8.7 fL. (D) ITGA2B expression is higher in septic patients who did not survive. Quartiles of ITGA2B mRNA expression were determined a priori and assigned as follows: Q1, <2.56 fold-change; Q2 between 2.56 and 4.09 fold-change; Q3, between 4.09 and 6.95 fold-change; Q4, >6.95 fold-change. Analyses in panels C-D were performed by analysis of variance. (E) Kaplan-Meier curve of mortality in mice following CLP (n = 20 mice). (F) Whole blood (2 μL) sampled from the tail vein 24 hours following CLP. Platelet αIIb surface protein was assessed 24 hours following CLP by flow cytometry, stratified for mice that subsequently did not survive (dead) or survived (alive) CLP. MFI, mean fluorescence intensity.
Increased ITGA2B and αIIb expression in sepsis is associated with both clinical and experimentally induced sepsis-related mortality. (A) In septic patients (n = 94), sepsis-related mortality is more than threefold higher in patients with thrombocytopenia at ICU admission (day 1). (B) Platelet counts in septic patients are inversely associated with MPV. (C) ITGA2B expression is significantly higher in newly released platelets (ie, platelets with a larger MPV) from septic patients. Quartiles for MPV were determined a priori and assigned as follows: quartile 1 (Q1), MPV <7.5 fL; Q2, MPV between 7.6 and 8.1 fL; Q3, MPV between 8.2 and 8.7 fL; Q4, MPV >8.7 fL. (D) ITGA2B expression is higher in septic patients who did not survive. Quartiles of ITGA2B mRNA expression were determined a priori and assigned as follows: Q1, <2.56 fold-change; Q2 between 2.56 and 4.09 fold-change; Q3, between 4.09 and 6.95 fold-change; Q4, >6.95 fold-change. Analyses in panels C-D were performed by analysis of variance. (E) Kaplan-Meier curve of mortality in mice following CLP (n = 20 mice). (F) Whole blood (2 μL) sampled from the tail vein 24 hours following CLP. Platelet αIIb surface protein was assessed 24 hours following CLP by flow cytometry, stratified for mice that subsequently did not survive (dead) or survived (alive) CLP. MFI, mean fluorescence intensity.
Discussion
Platelets are emerging as a potential therapeutic target in sepsis as a strategy to mitigate injurious thrombotic and inflammatory host responses.30 Acute alterations in rapid platelet functional responses (eg, integrin activation, P-selectin surface translocation, and platelet-leukocyte aggregate formation) are common in sepsis and, in some settings, correlate with adverse clinical outcomes.34,35 Relevant preclinical models of sepsis are critical for biological discovery and the development of new therapies. Studies directly comparing platelet reprogramming events (eg, changes in the repertoire and expression of mRNAs and proteins) in sepsis patients and experimental models of sepsis are absent in the field. Here, we coupled next-generation RNA-seq with ribosome footprint profiling,16 an innovative methodology to studying mRNA translation events, in platelets from septic patients and murine models of sepsis. To the best of our knowledge, this is the first study to globally profile the transcriptional and translational landscape of platelets during sepsis and perform comparative analyses between human and murine gene expression in platelets during sepsis.
We found that sepsis significantly altered the expression of numerous transcripts in platelets, with the majority (64%) being upregulated. Changes in gene expression were highly significantly correlated between septic patients and mice (ρ = 0.43, P = 3.2 × 10−285). Utilizing a strategy to identify significant, differentially expressed orthologs in human and murine platelets during sepsis, we identified >500 transcripts that were altered in both species concordantly in any direction (eg, upregulated or downregulated). Intriguingly, we found the strongest correlations between septic patients and mice were for translational events (ρ = 0.65, P = 1.81−90), suggesting that regulation of protein synthesis was tightly coordinated between species. These data indicate that for hundreds of genes, the CLP model of murine sepsis may have clinical relevance for examining sepsis-induced transcriptional and translational changes in platelets. The provided list of concordant changes in human and murine platelets during sepsis (supplemental Table 1) may serve as a resource for other investigators in the field. These data may also help fill current knowledge gaps on cellular changes occurring during the course of sepsis and help guide therapeutic efforts targeting pathophysiological responses in sepsis.
Our finding that the expression of many platelet mRNAs is increased in sepsis is consistent with microarray-based studies of platelets during murine sepsis. Using the CLP model, other investigators identified 56 genes that were differentially regulated in platelets isolated 24 to 48 hours following sepsis onset.36 We did not see substantial overlap between differentially expressed genes in our murine studies and these 56 genes, although differences in experimental conditions, platelet isolation techniques, and sequencing platforms (eg, microarray vs RNA-seq) confound direct comparisons.
Utilizing clinical (human) and experimental (murine) samples, we tracked the degree and timing of expression and function of ITGA2B (which encodes for the αIIb subunit of integrin αIIbβ3). We focused on ITGA2B, as it is an abundant, highly upregulated transcript in sepsis, is specifically expressed in platelets and megakaryocytes, is conserved between human and murine species, and has been investigated as a therapeutic target in sepsis. By multiple complementary approaches, we identified that ITGA2B mRNA was significantly upregulated in human and murine platelets during sepsis, with coordinate increases in the de novo synthesis of αIIb protein. Our findings suggest that increased Itga2b in platelets during sepsis was driven by increased trafficking of Itga2b into platelets from their parent cell, the megakaryocyte. These findings parallel previous murine based studies of granzyme B in sepsis, a proinflammatory gene also found to be upregulated in megakaryocytes following sepsis with subsequent investment in platelets.36 In septic patients, basal activation of integrin αIIbβ3 was significantly increased. We also identified that higher expression of integrin αIIb was associated with sepsis-induced mortality both in mice and in septic patients.
Given that dysregulated thrombosis and vascular complications are common in sepsis, integrin αIIbβ3 has been studied as a therapeutic target in sepsis. Studies in murine and baboon models of sepsis demonstrate that pharmacologic inhibition of αIIbβ3 improves parameters of endothelial function, organ failure, and mortality.25 While there have not been any clinical trials treating septic patients with integrin αIIbβ3 inhibitors, observational studies in sepsis suggest that antiplatelet agents (some of which block integrin αIIbβ3) are associated with improved clinical outcomes in some settings.37,38
Our findings also demonstrate that protein synthesis is increased in human and murine platelets during sepsis. We observed increases in global protein synthesis (as evidenced by 35S-methionine labeling and ribosome footprint profiling) and also dissected in detail the de novo synthesis of αIIb protein. Although anucleate, platelets have been recognized for many years to participate in protein synthetic events, and some of these occur in signal-dependent fashion in response to specific inflammatory agonists.39,40 Our study builds upon and extends these prior reports41,-43 by showing that sepsis, a systemic inflammatory syndrome, also triggers enhanced translational responses in human and murine platelets.
The MPV did not differ between septic patients and healthy controls, suggesting that increased ITGA2B mRNA expression in sepsis was not merely reflecting an increased pool of circulating younger and larger platelets. Nevertheless, in septic patients, our findings do suggest that ITGA2B mRNA is higher in newly released or younger platelets. This is consistent with published studies indicating that younger platelets in general contain more RNA (eg, reticulated platelets) that are associated with an increase in size.43,44 However, our studies were not designed to look specifically at transcriptional or translational responses as a function of platelet age in the circulation. We also focused on comparative analyses between septic patients and healthy donors, rather than attempting to correlate findings with variables of comorbid medical conditions and prescribed medications, which was not feasible to do and could not be extrapolated to experimental models of sepsis.
Strengths of our study include our parallel experiments in septic patients and murine CLP, integration of both RNA-seq and ribosome footprint profiling, and investigation in the magnitude of expression, migration, and functional significance of αIIβ in sepsis. We confirmed the de novo synthesis of αIIβ using parallel techniques of ribosomal footprint profiling, 35S-methionine pull-down, and in situ translation assays. While we observed increased mortality in septic patients with high platelet ITGA2B expression, these results are limited by the heterogeneity inherent in sepsis and potential confounding with regards to dynamic variation in platelet volume and turnover during sepsis.
Findings from our in vivo models recapitulated observations made in septic patients, although we cannot exclude the possibility of a survival bias in our murine CLP model. We also did not investigate other models of murine sepsis, which may result in other changes in gene expression and/or platelet functional responses. Differences between human and mouse transcriptional and translational response may also be influenced by type of infection and severity of disease. Additionally, for other mRNAs with increased ribosomal occupancy, we cannot exclude the possibility that this pattern was due to decreased termination and subunit dissociation, rather than increased initiation of translation.
Finally, while we identified an association between increased ITGA2B transcription and translation and mortality in both septic patients and mice, the functional consequences of increased ITGA2B transcription and translation remain unidentified. However, our finding of an increased number of copies of the activated conformation of integrin αIIb on the platelet surface is consistent with, and perhaps suggestive of, an increase in total integrin expression. Clear insight into the functional significance of increased ITGA2B mRNA and translation of its protein integrin subunit αIIb will be the result of future inquiry and studies.
In conclusion, our findings provide novel evidence that the platelet transcriptional and translational landscape is altered during clinical and experimental sepsis. These changes in gene expression are accompanied by upregulation of protein synthetic events and impact host responses in sepsis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Diana Lim for her creativity and expertise in figure preparation and Toni Blair for her editorial assistance.
This work was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute grants HL112311, HL126547 [M.T.R.], and R37HL044525 [G.A.Z.]; National Institute on Aging grant AG048022 [M.T.R.]; National Institute of General Medical Sciences grant GM103806 [J.W.R.]; and National Institute of Neurological Disorders and Stroke grant U10NS086606 [R.A.C.]) as well as by the German Research Foundation (Research Fellowship KR 4945/1-1) (K.K.). This material is the result of work supported with resources and the use of facilities at the George E. Wahlen Veterans Affairs Medical Center, Salt Lake City, UT. E.A.M. is supported by the University of Utah VPCAT Scholars Program and a Department of Internal Medicine SEED Grant. Research reported in this publication was supported by the National Center for Research Resources of the National Institutes of Health under award number 1S10RR026802-01. The work was also supported by the Health Sciences Center Cores at the University of Utah.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: E.A.M., R.A.C., H.S., J.W.R., B.K.M., Y.K., N.D.T., K.K., L.G., A.S.E., S.M.B., S.J.B., C.K.G., E.S.H., R.P., G.A.Z., A.S.W., and M.T.R. designed and performed experiments; E.A.M., J.W.R., A.S.W., and M.T.R. analyzed results and made the figures; E.A.M. and M.T.R. wrote the manuscript; and all authors reviewed and critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elizabeth A. Middleton, Molecular Medicine Program, Department of Internal Medicine, Eccles Institute of Human Genetics, Room 4220A, University of Utah, Salt Lake City, UT 84112; e-mail: elizabeth.middleton@hsc.utah.edu.