


Abstract
Filamins (FLNs) are large dimeric actin-binding proteins that regulate actin cytoskeleton remodeling. In addition, FLNs serve as scaffolds for signaling proteins, such as tyrosine kinases, GTPases, or phosphatases, as well as for adhesive receptors, such as integrins. Thus, they connect adhesive receptors to signaling pathways and to cytoskeleton. There are 3 isoforms of FLN (filamin a [FLNa], FLNb, FLNc) that originate from 3 homologous genes. FLNa has been the recent focus of attention because its mutations are responsible for a wide spectrum of defects called filaminopathies A, affecting brain (peri-ventricular nodular heterotopia), heart (valve defect), skeleton, gastrointestinal tract, and, more recently, the megakaryocytic lineage. This review will focus on the physiological and pathological roles of FLNa in platelets. Indeed, FLNa mutations alter platelet production from their bone marrow precursors, the megakaryocytes, yielding giant platelets in reduced numbers (macrothrombocytopenia). In platelets per se, FLNa mutations may lead to impaired αIIbβ3 integrin activation or in contrast, increased αIIbβ3 activation, potentially enhancing the risk of thrombosis. Experimental work delineating the interaction of FLNa with its platelet partners, including αIIbβ3, the von Willebrand factor receptor GPIb-IX-V, the tyrosine kinase Syk, and the signaling pathway of the collagen receptor GPVI, will also be reviewed.
Introduction
Platelets are produced by megakaryocyte (MK) cytoplasmic fragmentation and are critical in bleeding arrest. They adhere to vascular lesions, rapidly recruiting additional platelets until blood stops leaking. Excessive platelet accumulation at sites of atherosclerotic plaque rupture leads to arterial thrombus formation and acute myocardial infarction, sudden death, or ischemic stroke. Conversely, defective platelets lead to unstoppable hemorrhage. In the last decades, the challenge has been to identify molecular actors involved in platelet functions to identify potential drug targets. Recently, protein defects associated with signaling pathway dysfunction have been identified, such as filaminopathy A, a defect in filamin A (FLNa). FLNa is a large dimeric actin-binding protein that stabilizes actin filament networks; it was recently found to be involved in platelet production and platelet activation. This review is focused on the structure and functions of FLNa during platelet activation and megakaryocytopoiesis. We also discuss the molecular mechanisms involving FLNa in platelet activation. Finally, we analyze the hemorrhagic or thrombotic consequences of FLNa mutations in patients.
FLNa structure
FLNs in mammals are a family of 3 members, FLNa, FLNb, and FLNc, originating from 3 distinct genes (FLNA, FLNB, FLNC) that are highly conserved. Human FLNA is located on the X chromosome, whereas human FLNB and FLNC are located on chromosomes 3 and 7, respectively. FLN protein isoforms exhibit high sequence identity (70%) except for the 2 hinge regions.
FLNa monomers (280 kDa, 2646 aa) assemble as dimers in a V-shaped structure. The N- terminal region of each monomer is an actin-binding domain (ABD) consisting of 2 calponin homology (CH) sequences (CH1 and CH2) followed by a rod segment consisting of 24 immunoglobulin (Ig)-like repeats of 96-aa residues that adopt an Ig-like fold (Ig repeats) (Figure 1). The 24 Ig repeats are composed of 7 β-strands (A-G).1 Two calpain-sensitive hinge domains separate the 24 β pleated sheet Ig repeats into rod-1 (repeats 1-15; 58 nm) and rod-2 (repeats 16-23; 19 nm) and represent cleavage sites for Ca2+-dependent protease calpain.2 Rod-1 Ig repeats (9-15) also bind F-actin but with a lower affinity than the ABD, adding intrinsic flexibility to actin networks upon mechanical stress. Although FLNa rod-1 binds only a small number of partners, Ig repeats (16-24) in rod-2 interact with most FLNa partners (>90), including membrane receptors (GPIbα and integrin β3), signaling proteins (GTPase-related proteins), and transcription factors, conferring an important signaling scaffold role in a large variety of cellular processes.3 However, rod-2 does not bind F-actin. Rod-2 forms a structure that is more compact and more globular than rod-1 as the result of interactions between repeat pairs (16-17, 18-19, and 20-21). For example, strand A of Ig repeat 20 is associated with the β strands C and D of Ig repeat 21, thereby obstructing the integrin binding site.4 This is an autoinhibition mechanism that limits accessibility to the FLNa integrin-binding site, which may represent a general mechanism regulating FLNa–partner interactions. In response to mechanical forces, FLNa Ig-repeat domains undergo conformational changes, allowing the exposure of binding sites for new interactions. Thus, in platelets, FLNa may participate in mechanotransduction converting mechanical forces into signaling events. For example, disruption of Ig repeats 20-21 interaction (requiring mechanotransduction) enhances FLNa β-strand C and D binding to integrin β-tails.5 The C-terminal region (repeat 24) mediates dimerization into a V-shaped flexible structure that results in F-actin perpendicular branching that is essential for mechanosensory functions,6 although other domains may participate in dimerization.7 Finally, repeat 24 also interacts with other FLNa partners, such as the small GTPases RalA, Rho,8 or Cdc42,9 involved in particular in actin cytoskeleton regulation and the endoplasmic reticulum (ER) calcium sensor stromal interaction molecule 1 (STIM1), regulating Ca2+ influx through Orai-1.10
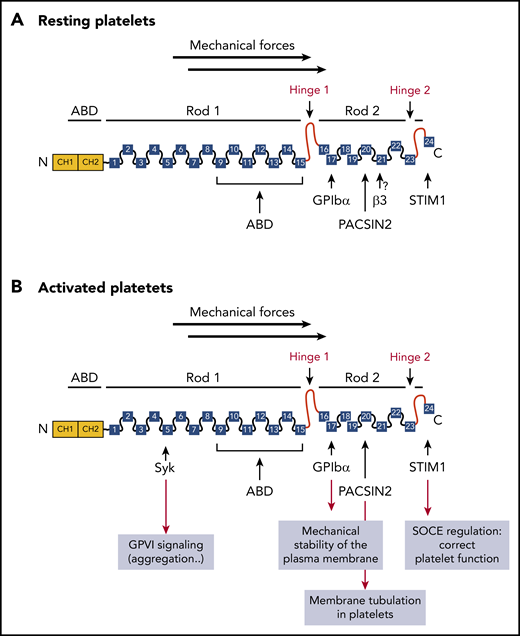
Monomeric structure of FLNa and partners in platelets. The amino-terminal ABD contains 2 CH domains (CH1 and CH2), followed by 24 Ig repeats, probably folded into antiparallel β-sheets. Two hinge domains separate the 24 Ig repeats into 2 rod domains (hinge 1: between Ig repeats 15 and 16 and hinge 2 between Ig repeats 23 and 24). Ig repeats 9 to 15 in rod-1 facilitate F-actin binding, whereas Ig repeats 16 to 23 interact with different partners. Dimerization occurs through Ig repeat 24. (A) FLNa interacts with the platelet receptor GPIbα through the Ig repeat 17, as well as with the recently described partners PACSIN2 and STIM1 in resting platelets. The interaction of FLNa with αIIbβ3 through the Ig repeat 21 was not demonstrated. (B) After platelet activation, FLNa interacts with the platelet receptors GPIbα and with the tyrosine kinase Syk involved in GPVI functions through the Ig repeats 17 and 5, respectively, as well as with PACSIN2 and STIM1. SOCE, store-operated calcium entry.
Monomeric structure of FLNa and partners in platelets. The amino-terminal ABD contains 2 CH domains (CH1 and CH2), followed by 24 Ig repeats, probably folded into antiparallel β-sheets. Two hinge domains separate the 24 Ig repeats into 2 rod domains (hinge 1: between Ig repeats 15 and 16 and hinge 2 between Ig repeats 23 and 24). Ig repeats 9 to 15 in rod-1 facilitate F-actin binding, whereas Ig repeats 16 to 23 interact with different partners. Dimerization occurs through Ig repeat 24. (A) FLNa interacts with the platelet receptor GPIbα through the Ig repeat 17, as well as with the recently described partners PACSIN2 and STIM1 in resting platelets. The interaction of FLNa with αIIbβ3 through the Ig repeat 21 was not demonstrated. (B) After platelet activation, FLNa interacts with the platelet receptors GPIbα and with the tyrosine kinase Syk involved in GPVI functions through the Ig repeats 17 and 5, respectively, as well as with PACSIN2 and STIM1. SOCE, store-operated calcium entry.
Regulation of FLNa interaction with its partners
FLNa interaction with its partners is regulated by phosphorylation, proteolysis, mechanical forces, and competition between partners. Several kinases, including cyclic adenosine monophosphate (cAMP)–kinase, protein kinase C, and Ca2+/calmodulin-dependent protein kinase II, phosphorylate FLNa on S2152.11-13 The physiological roles of this phosphorylation remain largely uncharacterized. FLNa phosphorylation on S2152 by the cAMP-dependent protein kinase (protein kinase A [PKA]) was reported to stabilize and protect FLNa against proteolysis by calpain.14 Indeed, we recently described a new gene mutation in PRKACG encoding the γ regulatory subunit of PKA, which was associated with a macrothrombocytopenia and with a functional defect in PKA activity, leading to a decrease in FLNa phosphorylation at S2152 and to FLNa degradation.15 Phosphorylation may also regulate FLNa interaction with GTP-binding proteins16 or with actin filaments,11 and it may regulate the role of FLNa in integrin activation.17
Mechanical strain is yet another essential actor regulating FLNa interaction with its partners.18,19 This mechanical stress-dependent mechanism may result from the lift of an autoinhibitory pairing of Ig domains (Ig 20-21, 18-19, 16-17), limiting binding sites’ accessibility to FLNa partners. The pair Ig 20-21 was the most studied. Recent in vitro studies of mechanically strained FLNa–cross-linked actin networks have provided further support for a potential force-sensing role for FLNa.18 Ehrlicher et al examined the effect of mechanical strain on FLNa’s interactions with 2 rod-2 binding partners: cytoplasmic β-tail integrin and FilGAP, a GTPase specific for Rac.18 In this model, strain increased β-integrin binding to FLNa, whereas it caused the dissociation of FilGAP from FLNa, indicating that FLNa rod-2 is highly flexible and that physiological forces are sufficient to expose new cryptic sites. The dynamic force sensing of FLNa, also revealed in single-molecule experiments, was shown to be a highly dynamic process shifting the autoinhibited domain pair Ig 20-21 toward the increased binding affinity of β-integrin to IgFLNa 21.20 It remains to be established in resting platelets whether and how the interaction of FLNa with β3 occurs. To answer this question, the presence or not of the autoinhibitory Ig 20-21 domain pairing must be determined in platelets. Finally, competition is an important regulatory mechanism of FLNa interaction with its partners. For example, competition between FLNa and talin or kindlin-3 has been extensively studied with respect to αIIbβ3 integrin and will be discussed in "FLNa partners in platelets (subsection "The integrin αIIbβ3").
FLNA mutations are associated with a wide spectrum of genetic diseases
FLNA mutations cause a wide spectrum of rare diseases, including skeletal dysplasia, neuronal migration abnormality putatively linked to intellectual disability, cardiovascular malformation, myofibrillar myopathy, and intestinal malrotation and obstruction (Table 1). FLNA mutations are distributed throughout the entire FLNA gene and give rise to abnormal messenger RNA splicing or protein truncations.21,22 These disorders are called filaminopathies A; the most frequent disease is periventricular nodular heterotopia (PVNH), which is characterized by a neuronal migration defect leading to ectopic accumulation of neurons in nodules lining cerebral ventricle margins.21-23 PVNH is predominantly observed in heterozygous females exhibiting loss-of-function mutations.22,24 Filaminopathy A in males is most frequently associated with perinatal lethality, with only rare hemizygous males surviving postbirth.21,24-26 Most PVNH mutations disrupt the FLNA reading frame and completely ablate FLNa expression, with only a small number of missense mutations identified in the ABD (CH1). In other cases, the PVNH phenotype is associated with Ehlers-Danlos syndrome,27 with vascular defects including aortic valve insufficiency and patent ductus arteriosus,22 hydrocephalus,28 frontonasal malformation,29 nephrosis,30 or congenital intestinal pseudo-obstruction.26,31
FLNA mutations are also associated with skeleton alterations, such as frontometaphyseal dysplasia, Melnick-Needles syndrome, and otopalatodigital syndrome type 1 and type 2 (OPD1 and OPD2).32 OPD1 syndrome is characterized by facial malformation and generalized bone dysplasia.33 OPD2 syndrome is a more severe disorder, associated with mental retardation.34 In contrast to PVNH, mutations leading to OPD are primarily localized in specific domains. The actin binding domain (CH2) may harbor gain-of-function mutations with increased actin-binding affinity. Rod domain repeats 3, 10, and 14/15 are the target of many missense mutations affecting partner binding and, consequently, FLNa functions.32,35,36 Likewise, recurrent FLNA mutations in the ABD (p.E254K) associated with OPD2 and in the Ig10 (p.S1199L) associated with Melnick-Needles syndrome have been observed in unrelated families,32,36,37 strongly suggesting that different FLNa functions and/or partners are involved in bone and brain development and remain to be identified.
Finally, FLNA was identified as the first gene responsible for myxomatous valvular dystrophy, and 4 mutations have been clearly identified38,39 : 3 missense point mutations within Ig repeats 1, 4, and 5 and a deletion encompassing Ig repeats 5 to 7. Interestingly all mutations are predicted to impair binding of FLNa partners by alteration of the β-strand organization.
FLNa in hemostasis
Until 2010, thrombocytopenia and hemorrhages have only occasionally been described in PVNH patients.22 The role of FLNa in platelets and MKs has been unraveled first in mice lacking FLNa40,41 and in mouse embryonic stem cells (ESCs)42 or in patients carrying FLNA mutations.26,27,43-45 MK-platelet lineage–specific FlnA−/− mice exhibit increased tail bleeding and severe macrothrombocytopenia due to accelerated platelet clearance.41 FlnA-null MKs prematurely release large, fragile, and vesiculate platelets that are cleared from the circulation by macrophages.41 Impaired megakaryopoiesis was also noted in 2 female patients, 1 with PVNH and the other not suffering from filaminopathy, but both exhibited hemorrhages and macrothrombocytopenia (Table 2).45 Their MKs were morphologically abnormal, including enlarged α granules, abnormal cytoplasm fragmentation, and impaired proplatelet formation, associated with uneven FLNa distribution. An altered demarcation membrane system (DMS) was also observed in patients,45 as well as FlnA-null MKs,46 the likely result of an impaired FlnA–PACSIN2 interaction, which regulates membrane tubulation in MKs and platelets and DMS formation, at least in mice.46
More recently, induced pluripotent stem cells (iPSCs) derived from heterozygous female patients with PVNH43,44 were used to assess variability in patients’ platelet counts.43 In fact, 2 platelet populations differing in size were observed in these heterozygous female patients. Moreover, random X inactivation during hematopoiesis suggests the generation of 2 MK progenitor populations exclusively expressing the wild-type (wt) or the null X-linked FLNA allele in heterozygous PVNH female patients. In fact, the reprogramming of FLNa-wt and FLNa-null clones isolated from each female patient showed no difference in MK differentiation, up to proplatelet formation, which was highly defective in the iPSC-derived FLNA-null MKs.43
Platelets predominantly express FLNa. Mouse platelets lacking FlnA are large, but discoid, with impaired actin membrane attachment and GPIbα expression.40 The platelet morphology studied in 2 PVNH female patients exhibiting FLNA premature termination mutations and a female patient with isolated macrothrombocytopenia and an FLNA missense mutation45 showed 2 platelet populations. Some platelets were normal, whereas others were giant with an abnormal granule distribution.27,44,45 Note that, in contrast to murine platelets, enlarged human platelets are round, and no GPIbα decrease or degradation was observed, suggesting that the GPIbα-FLNa-actin axis is regulated differently in mice and in humans. In human platelets, FLNa distribution was irregular, with low or no FLNa in giant platelets, whereas the FLNa level was normal in normal-sized platelets. Surprisingly, in patients with putatively truncated FLNa, full-length FLNa was detectable but at variable levels. In these patients, altered platelet functions correlated with low full-length platelet FLNa levels. Moreover, platelet count correlated with the wt FLNa level, with low counts associated with low FLNa levels and normal counts associated with normal FLNa levels.44
A different question is raised by missense FLNa mutants with mutations not affecting expression or stability, thereby presumably expressed in platelets at similar levels as wt FLNa. Could these FLNa mutants exhibit a dominant alteration in platelet function leading to thrombosis or hemorrhage? The first case was a missense FLNA mutation (pGlu1803Lys) in 1 heterozygous female patient exhibiting a gain-of-platelet function with increased adhesion on von Willebrand factor (VWF) in conditions of pathological shears.44 This effect may be related to the location of the mutation within Ig repeat 16, next to Ig repeat 17, the FLNa–GPIb interaction domain.47 One possibility is that this mutation disrupts the Ig repeats 16/17 autoinhibitory pairing, facilitating GPIb binding, as suggested by the FLNa model of Ehrlicher et al.18 Another platelet gain-of-function effect was reported in a hemizygous male patient (therefore carrying a single X-linked FLNA allele) with an extended C-terminal region due to a stop codon mutation.26 Accordingly, the platelets of this hemizygous patient expressed only the mutant FLNa protein. Despite normal αIIbβ3 expression, αIIbβ3 exhibited enhanced ligand binding (twice the normal values), exposing this patient to a potential thrombotic risk. Because mutant FLNa seemed to bind β3 on resting platelets, we postulate that the gain-of-function effect is likely to be due to an easier release of mutant FLNa from β3 than normal, facilitating talin recruitment and overactivation of αIIbβ3. Thus, this is the first observation in humans confirming a downregulatory role for FLNa in αIIbβ3 activation, a key feature in platelet physiology and pathology.
FLNa partners in platelets
Because FLNa interacts with several receptors (GPIbα and αIIbβ3 integrin) essential to platelet adhesion and aggregation, and it binds to signaling proteins, such as Syk, involved in GPVI functions, and STIM1 (Figure 1), filaminopathy A patients and the corresponding mouse models provide opportunities to examine the role of FLNa in platelet functions and signaling.
GPIb-IX-V
GPIb-IX-V, a platelet-specific membrane glycoprotein, mediates adhesion of platelets to blood vessel wall injury sites by binding to VWF immobilized on the subendothelial collagen matrix. FLNa constitutively interacts with GPIb-IX-V and positively modulates VWF receptor function.48,49 This interaction involves FLNa repeat 17 and GPIbα subunit cytoplasmic tail (aa 556-577).47,50,51 Other Ig repeats (4, 9, 12, 17, 19, 21, and 23) may also bind GPIbα, but double-missense mutations of Ig repeat 17 were sufficient to completely abolish FLNa–GPIb-IX-V interaction.1,51 Phe568 and Trp570, within the hydrophobic FLNa binding site of the GPIbα cytoplasmic tail,48 appear to be essential to strengthen GPIb-IX-V anchorage to the membrane skeleton and cell adhesion to VWF under high shear.48 This was later confirmed with a GPIbα knock-in mouse model in which GPIbα cytoplasmic tail was deleted and replaced by the reporter protein β-galactosidase.52 Blunting GPIbα–FLNa interaction led to defective adhesion, unstable membrane tethers, and loss of membrane integrity at pathological shear rates, indicating that FLNa–GPIbα linkage ensures mechanical stability of the platelet plasma membrane.
FLNa has also been shown to be required for GPIbα trafficking. FLNa involvement in GPIb trafficking to the plasma membrane was initially observed in FLNa-defective melanoma M1 cells,53 in which GPIb-IX reached the cell surface only when coexpressed with FLNa; GPIb-IX remained in the cytoplasm in the absence of FLNa. FLNa–GPIbα interaction was proposed to regulate posttranslational GPIb-IX-V assembly and trafficking; the binding of FLNa to GPIbα would trigger GPIb-IX-V ER exit, allowing post-ER trafficking.54 Moreover, the FLNa expression level also seems to be important for GPIbα trafficking.43 This mechanism could explain the apparent discrepancy between low FLNa levels together with normal or increased levels of membrane GPIb-IX-V in PVNH patients’ platelets, and no FlnA together with low levels of GPIb-IX-V in FlnA-null mouse platelets.40,41 Of note, 20% normal FLNa in heterozygous patients’ platelets seemed to be sufficient for normal GPIb-IX-V platelet surface expression.44
The integrin αIIbβ3
Extensive studies have been carried out with regard to the role of FLNa in the regulation of integrin activation. For example, FLNa was shown to interact with β7 tail integrin.5 However, the role of the αIIbβ3–FLNa interaction in platelets remains unclear. A recent elegant structural study proposed a new model whereby Ig repeat 21 (and possibly Ig repeats 9, 12, 17, and 19) of FLNa clasps together αIIb and β3 cytoplasmic tails (CTs), thereby stabilizing αIIbβ3 in an inactive state and preventing spontaneous activation of αIIbβ3.55 In this model, FLNa was proposed to prevent talin interaction with β3 and to mediate the retention of the integrin in a resting state. However, FLNa–β3 interaction was never demonstrated in intact resting platelets. This finding was consistent with the observation that basal αIIbβ3 activation was not observed in resting platelets from the patient with a gain-of-platelet function26 or from FlnA-null mice,40 despite normal and increased expression levels of αIIbβ3, respectively. The apparent discrepancy between intact platelets and the recent model proposed by Liu et al55 could come from the model that those investigators used, which consisted of an Ig repeat 21 peptide alone (ie, without the autoinhibiting Ig repeat 20). Thus, this model would allow free accessibility to the FLNa integrin-binding site. It remains to be clarified whether Ig repeat 21 is inhibited by Ig repeat 20 in platelets under physiological conditions and whether this inhibition could be eradicated by high shear conditions.
The molecular basis of the dynamic equilibrium between resting and activated αIIbβ3 is not completely understood. Resting platelets exhibit low-affinity αIIbβ3 displaying minimal ligand binding. After platelet activation, αIIbβ3 binds ligands with a high affinity and avidity. αIIbβ3 high-affinity transition is regulated by talin, kindlin, and FLNa direct interaction with β3 CT. Talin and kindlins (kindlin-3 in hematopoietic cells) are known integrin activators, whereas FLNa is a β1 and β3 integrin inhibitor.56,57 Altered integrin activation is observed in talin-null platelets or after kindlin overexpression or knockout.58-61 Likewise, αIIbβ3 activation was impaired in knock-in mice expressing a kindlin-3 mutant disrupting kindlin-3–αIIbβ3 interaction.62 Kindlin-3 and talin seem to synergistically coactivate αIIbβ360,63 but with distinct roles: talin modulates αIIbβ3 affinity, whereas kindlin-3 modulates clustering and avidity.64 FLNa binds to β3 CT residues 747 to 755 overlapping with aa 739 to 750 and aa 752 to 759, which are talin and kindlin-3 binding sites, respectively. This supports the view that talin and kindlin-3 compete with FLNa for β3 CT interaction.57,65-67 The model proposes that platelet activation induces FLNa release from its constitutive β3 CT binding site, allowing talin and kindlin-3 β3 recruitment, and it suggests that FLNa would be capable of dissociating from β3 under platelet activation. In the new model proposed by Liu et al,55 FLNa would prevent talin and kindlin-3 binding to β3 CT, as well as membrane insertion of the membrane-proximal region of β CT. FLNa release would allow talin and kindlin-3 β3 CT binding and also let the αIIbβ3 CT membrane-proximal region insert into the membrane and undergo conformational rearrangement and, thus, expose the ligand binding site. Thus, FLNa would represent a double lock for αIIbβ3 activation.
What is the regulatory mechanism for FLNa dissociation from αIIbβ3? Migfilin, another FLNa partner reported to regulate integrin activation, has been proposed to play a role in this mechanism. Overexpression of migfilin significantly enhanced integrin activation,56 and permeable migfilin peptides in platelets were able to induce extensive αIIbβ3 activation. Structural analysis of the migfilin-FLNa complex identified Ig repeat 21 as the preferential migfilin binding site on FLNa (and to a lesser extent, Ig repeats 19 and 22),68 whereas, reciprocally, FLNa bound the migfilin N-terminal domain.56 Accordingly, a model was proposed whereby migfilin binding to FLNa would lead to FLNa release from β CT, thus leaving the β CT binding sites free for interaction with talin and kindlin-3.56,57,68 However, if this model is attractive, it was not demonstrated in platelets. Conflicting results have been published regarding the presence or absence of migfilin in platelets.57,69 Likewise, in human platelets, although FLNa and β3 copy numbers match (88 000 and 90 000 molecules, respectively), migfilin was not detected in proteomics analyses of human platelets.70 Together with the fact that only low levels of migfilin messenger RNA and protein have been reported,57 these data are inconsistent with a role of migfilin in displacing FLNa from β3 CT. Other unidentified candidates could play such a role. Alternatively, FLNa-β3 dissociation could also be the result of an FLNa conformational change that occurs during platelet activation, leading to a decreased FLNa affinity for β3 CT and to its dissociation from β3 (Figure 2).
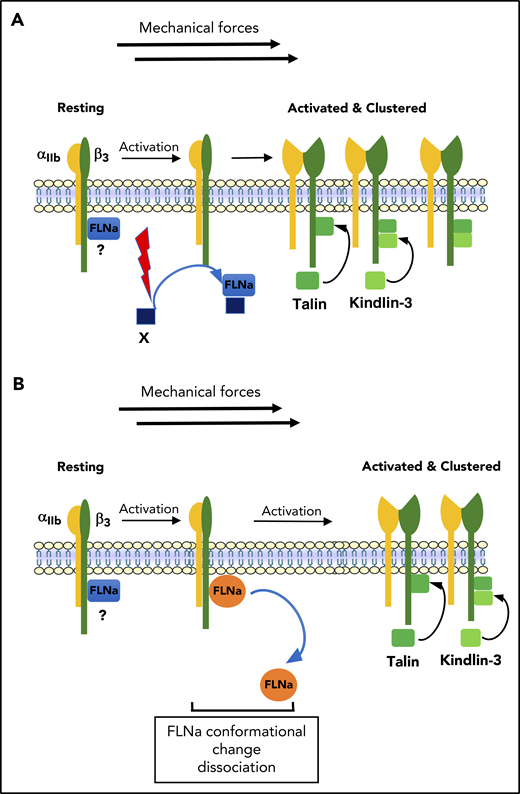
Model of the regulation of αIIbβ3 activation. (A) In resting platelets, FLNa is constitutively associated with αIIbβ3 (β3 CT) through its Ig repeat 21. After platelet activation, FLNa interacts with an unknown FLNa partner through Ig repeat 21, leading to FLNa release from βCT. In this model, FLNa-free βCT binding sites would now allow recruitment of talin and kindlin to βCT required for αIIbβ3 activation. (B) In resting platelets, FLNa is constitutively associated with αIIbβ3 (β3 CT) through its Ig repeat 21. The dissociation of FLNa from β3 could be the result of a conformational change in FLNa that occurred during platelet activation and in the presence of shear, leading to a decreased affinity and dissociation of FLNa from β3.
Model of the regulation of αIIbβ3 activation. (A) In resting platelets, FLNa is constitutively associated with αIIbβ3 (β3 CT) through its Ig repeat 21. After platelet activation, FLNa interacts with an unknown FLNa partner through Ig repeat 21, leading to FLNa release from βCT. In this model, FLNa-free βCT binding sites would now allow recruitment of talin and kindlin to βCT required for αIIbβ3 activation. (B) In resting platelets, FLNa is constitutively associated with αIIbβ3 (β3 CT) through its Ig repeat 21. The dissociation of FLNa from β3 could be the result of a conformational change in FLNa that occurred during platelet activation and in the presence of shear, leading to a decreased affinity and dissociation of FLNa from β3.
The negative regulation of αIIbβ3 activation by FLNa in platelets was clearly demonstrated in a rare case of a male patient exhibiting PVNH and congenital intestinal pseudo-obstruction. This male patient was hemizygous for a mutant FLNa exhibiting an extended (100-aa) C-terminal region due to the stop codon mutation and showed gain-of-platelet-function characteristics.26 The mutant FLNa was shown to augment αIIbβ3 activation by facilitating talin and kindlin-3 recruitment to αIIbβ3 integrin. This was not due to an absence of FLNa–β3 CT interaction, which appeared to be normal. The current hypotheses are that (1) the mutant FLNa, despite its ability to constitutively bind to β3 CT, is more easily released than wt FLNa from β3 CT and (2) because the mutation is next to the dimerization domain of FLNa, interference with dimerization may ease mutant FLNa release. Thus, this increased αIIbβ3 activation correlating with this FLNa mutation unravels an unsuspected set of potential mechanisms for FLNa-β3 CT dissociation that require further exploration. Finally, increased αIIbβ3 activation correlating with this FLNa mutation raises another question pertaining to the risk for spontaneous αIIbβ3 activation and, thus, for thrombosis, in this patient, particularly if he develops prothrombotic pathologies, such as atherosclerosis or diabetes mellitus.
The tyrosine kinase Syk
The tyrosine kinase Syk, which is involved in the signaling of immunoreceptor tyrosine-based activation motif and immunoreceptor tyrosine-based activation motif-like–mediated signal receptors GPVI and C-terminal lectin-like receptor 2, is also an important FLNa partner, as shown elegantly in MK-specific FlnA−/− mice; they exhibited altered collagen-induced platelet functions.40 Platelet adhesion under arterial shear was impaired, as were platelet aggregation and αIIbβ3 activation induced by GPVI-specific agonists, such as collagen-related peptide. Defective Syk phosphorylation confirmed that FlnA is essential for GPVI signaling. In fact, platelet FlnA contributes to Syk targeting to the cytoplasmic membrane leaflet. Interestingly, Syk was shown to bind FlnA rod-1 Ig repeat 5, contrasting with most other partners binding rod-2 Ig repeats 16 to 24.
FLNa was also shown to be involved in GPVI signaling in platelets from heterozygous female PVNH patients with macrothrombocytopenia and low FLNa levels.44 Abnormal responses to collagen, including aggregation, secretion, GPVI signaling, and thrombus formation under flow, were the consequence of low levels of wt FLNa (≤50%). Partial Syk activation impairment in patient platelets was probably the consequence of a defective Syk–FLNa association, which is required for Syk recruitment by GPVI-associated FcRγ chain.
New partners
PACSIN2
The adaptor PACSIN2 is 1 of the most abundant BAR/F-BAR proteins in platelets.70,71 The interaction of PACSIN2 with FLNa has recently been described in human platelets, and it involves FLNa Ig repeat 20 and the PACSIN2-BAR domain. This interaction regulates membrane tubulation in MKs and platelets and DMS formation in MKs.46
STIM1
Among the other signaling partners, the ER calcium sensor STIM1, which regulates store-operated calcium entry (SOCE), a major mechanism for Ca2+ influx, has recently been shown to interact with FLNa.10 This interaction is dependent on the phosphorylation of FLNa at Ser2152 by cAMP-dependent protein kinase and on the FLNa dimerization domain, and it seems to downregulate SOCE.10
Other partners
Among the 100 known partners of FLNa, only the FLNa/STIM1 and PACSIN2 interactions, described above, were demonstrated in platelets. Other candidates probably do interact with FLNa in platelets. For example, the GTPase FilGAP, which is expressed in all tissues, was shown to bind FLNa through Ig repeat 23.72 The dissociation of FilGAP from FLNa occurs when FLNa-actin networks are subjected to mechanical shear strains in vitro,18 which corresponds to fluid shear stress driven by blood flow. In this model, mechanical shear stress deforms actin filaments and, consequently, the 2 arms of FLNa, which reduces FilGAP-FLNa binding.72 It remains to be established whether this mechanism occurs in intact platelets and what its function is in the modulation of Rho and Rac activities.
FLNa binds constitutively to Rho family GTPases (Cdc42, RhoA, and Rac1) via Ig repeat 24. In MKs, the interaction of RhoA with FLNa was recently reported to be dependent on αIIbβ3 and to modulate proplatelet formation (see "FLNa in megakaryopoiesis").42
FLNa in megakaryopoiesis
Macrothrombocytopenia and defective proplatelet formation were observed in FLNa-knockout mouse models and mouse ESCs.41,42 In contrast to ESCs, in which inefficient MK differentiation was observed, knockout mice showed a marked increase in bone marrow and spleen MKs.
A recent original iPSC model, using reprogramming of peripheral blood CD34+ cells from 2 female patients harboring 2 intragenic deletions, showed that, in differentiated MKs, there was no defect in CD41+/CD42+ MK percentage or mean fluorescence intensity for αIIbβ3, GPIX, or GPIbα expression.43 These observations suggested that, in human MKs, trafficking to the cell surface of GPIbα or αIIbβ3 is independent of FLNa. In contrast, complete FLNa ablation in MKs was associated with defective proplatelet production.43 MK differentiation onto fibrinogen, thus involving αIIbβ3 engagement and presumably FLNa release from αIIbβ3, led to increased actomyosin contractility associated with inappropriate GTPase RhoA activation, resulting in defective platelet production. Thus, downregulation of RhoA activity by FLNa–αIIbβ3 interaction is required for proplatelet formation. This was confirmed by experiments overexpressing FLNa-deletion mutants unable to bind αIIbβ3 and RhoA (Figure 3). This is the first report proposing a molecular mechanism for macrothrombocytopenia involving FLNa.43 A defective FLNa–αIIbβ3 interaction may also explain thrombocytopenia in some patients with αIIb and β3 mutations.73,74

FLNa and platelet biogenesis. (A) αIIbβ3/FLNa interaction after adhesion to fibrinogen is crucial to keep RhoA inactive, allowing actomyosin reorganization and proplatelet formation. (B) GPIbα–FLNa interaction stabilizes platelets and regulates the size under shear. It cannot be excluded that this interaction is necessary for the regulation of GPIb downstream effectors RhoA/Cdc42 during transendothelial platelet biogenesis.
FLNa and platelet biogenesis. (A) αIIbβ3/FLNa interaction after adhesion to fibrinogen is crucial to keep RhoA inactive, allowing actomyosin reorganization and proplatelet formation. (B) GPIbα–FLNa interaction stabilizes platelets and regulates the size under shear. It cannot be excluded that this interaction is necessary for the regulation of GPIb downstream effectors RhoA/Cdc42 during transendothelial platelet biogenesis.
There is increasing evidence that GPIbα–FLNa interaction maintains platelet membrane mechanical stability at high shear rates, as well as regulates platelet size. Indeed, the abnormal architecture of (giant) platelets associated with the inherited deficiency in GPIb-IX-V (Bernard-Soulier syndrome) has been proposed to arise from the absence of GPIbα–FLNa interaction.75 This finding was supported by the presence of giant platelets in Bernard-Soulier syndrome patients with CT-truncated GPIbα or by the expression of transmembrane and cytoplasmic domains of GPIbα in GPIbα-deficient mice, which was sufficient to partially rescue platelet size.76 Similar to GPIbα-deficient mice, FLNa-deficient mice form giant platelets.77 Likewise, the population of giant platelets (20%) observed in female PVNH patients was associated with a large decrease in, or a total absence of, platelet FLNa, whereas normal-sized platelets were associated with the presence of wt FLNa.44,45 Finally, different levels of FLNa and GPIb expressed in cultured ESCs differentiated in MKs forming platelets confirmed their roles in the production of normal-sized platelets43 ; these indicate that platelet size is regulated by GPIbα–FLNa interaction, and macrothrombocytopenia may stem from a deficiency in FLNa or GPIbα. Finally, a model in which GPIbα controls MK localization and transendothelial MK migration via the regulation of the Rho GTPases RhoA and Cdc42 has recently been described.78 The role of FLNa in the transmission of signaling between GPIbα and Cdc42-RhoA remains to be explored.
The future
Recent studies on FLNa have changed our view of its role in platelets and MKs. The structure, the identification of its partners, the FLNa mutants, and, finally, the mouse models of FLNa deficiency have helped us to better understand the role of FLNa in hemostasis and megakaryocytopoiesis; however, they have also raised new questions.
Little is known about the molecular role of FLNa in megakaryocytopoiesis. We have seen that the absence of FLNa in MKs leads to their inability to produce proplatelets and that the downregulation of RhoA activity required for proplatelet formation is dependent on αIIbβ3–FLNa interaction.43 To investigate how RhoA is regulated, it will be interesting to identify which GAPs, GEFs (known to interact with FLNa), or other partners are able to modulate Rho activity. Another question relates to whether the RhoA pathway can be considered a potential new target for increasing platelet counts in patients with thrombocytopenia. Because the absence of FLNa in MKs leads to αIIbβ3-dependent overactivation of the RhoA pathway, the state of αIIbβ3 activation remains to be investigated. One surprise in the study was that deletion of the GPIb-binding domain did not alter proplatelet formation, in apparent contradiction with the reported effect of GPIb–FLNa interaction ablation causing giant platelets or thrombocytopenia in mice. One potential explanation is that data originated from an in vitro system lacking conditions required for final platelet generation, including shear. This could indicate that the role of FLNa–GPIb interaction in platelet formation is relevant at a late stage in platelet biogenesis. This possibility may be consistent with the observation that GPIbα was shown to coordinate MK polarization and transendothelial migration in vivo via the regulation of Cdc42 and RhoA activity.78
In platelets, the regulation of αIIbβ3 activation by FLNa remains unclear; the constitutive association of FLNa with β3 has not been shown to prevent spontaneous integrin activation, and the determinants of FLNa–αIIbβ3 interaction are not understood. Can FLNa–αIIbβ3 interaction be regulated by phosphorylation, similarly to FLNa-integrin β2 CT binding inhibition by Thr758 phosphorylation?79 Another important, but still elusive, question is if FLNa is associated with αIIbβ3, how does it dissociate from β3? We have seen that the cytoskeletal adaptor protein migfilin is not a good candidate for promoting FLNa release from β3 CT in platelets; thus, the molecular mechanism remains unknown.
The mechanisms by which different missense FLNa mutations in patients affect platelet production and/or functions is an important question, in particular because of the potential risk for hemorrhage or thrombosis. For example, the role of the dimerization domain (FLNa Ig repeat 24) in platelet production and activation, as well as the role of FLNa Ig repeat 21 in αIIbβ3 activation, must be elucidated. Addressing the hemostatic status of patients with filaminopathies A needs to be considered more systematically. The expected benefits are of a medical, physiopathological, and physiological nature. Moreover, it is quite possible that we will establish a relationship between the mutation type and its pathophysiological impact in the future. If such a correlation were made, subsequent monitoring of patients should be adjusted based on the mutation involved. With regard to the pathophysiology of the MK-platelet impact of filaminopathy A, we believe that future investigations should provide clues to the mechanism of thrombocytopenia.
Acknowledgments
The authors thank Paquita Nurden and Cecile V. Denis for insightful suggestions.
This work was supported by grants from INSERM and Fondation pour la Recherche Medicale (LPC20170637458).
Authorship
Contribution: J.-P.R., H.R., and M.B. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marijke Bryckaert, INSERM UMR_S 1176, Hôpital Bicêtre, 80 rue du Général Leclerc, 94276 Le Kremlin Bicêtre Cedex, France; e-mail: marijke.bryckaert@inserm.fr.


