

TO THE EDITOR:
Precursor B-cell acute lymphoblastic leukemia (B-ALL), a frequent malignancy in children, is characterized by recurrent primary numerical or structural alterations that provide specific molecular markers of the developing malignant clone.1 The most common alterations, accounting for >50% of all cases, are high hyperdiploidy (51-67 chromosomes) and the chromosomal translocation t(12;21) that generates the ETV6-RUNX1 (synonymous with TEL-AML1) fusion gene.2 Studies that use monozygotic twins concordant for leukemia, retrospective analyses of archived neonatal blood spots, and molecular screening of umbilical cord blood (UCB) have demonstrated that these subtypes of B-ALL frequently emerge before birth during fetal hematopoiesis.1,3-5 Both alterations are only mildly oncogenic and require secondary mutations for disease onset. This requirement partially explains the long latency of these leukemias, which emerge after birth at age 2 to 5 years (range, 1-15 years). Rarer B-ALL subtypes, characterized by KMT2A (MLL) translocations, are also generated in utero as evidenced by similar studies.6-8
In contrast, the translocation t(1;19), which generates the TCF3-PBX1 fusion gene, is currently assumed to arise after birth.9 This assumption is based on a study of 15 patients with TCF3-PBX1+ ALL whose neonatal blood spots were assessed for the presence of the patient-specific fusion breakpoint using polymerase chain reaction (PCR). Only 2 patients were possibly positive for the fusion in this study, and at that time, the introduction of nontemplate bases at the breakpoints and specific characteristics of the IGH/TCR rearrangements were interpreted as supportive for postnatal cell origin, although the cohort size was small.9 TCF3-PBX1+ B-ALL is frequent in childhood and accounts for ∼5% to 10% of cases,2,10-13 but there is a comparatively lower incidence in adolescence and adulthood (3%).2 The TCF3-PBX1 gene fusion, similar to ETV6-RUNX1, is only mildly oncogenic, requires secondary mutations for disease onset, and emerges usually at age 4 to 6 years.14,15 However, very young patients (younger than age 1 year) have also been diagnosed. A recent study reported on 4 t(1;19)-positive ALL patients age 10 to 22 months. This early disease onset leaves only a short time frame for secondary mutations to occur, hinting at a possible prenatal origin of the translocation.16 These findings prompted us to investigate the presence of TCF3-PBX1 translocations in newborns in detail.
Recently, we screened 1000 UCB samples and demonstrated that the ETV6-RUNX1 translocation is present in ∼5% of healthy newborns by using a DNA-based technique that we developed termed GIPFEL (genomic inverse PCR for ligated breakpoints).5,17 GIPFEL was designed to screen for translocations without knowing the exact breakpoint.17 GIPFEL detects 1 translocation-carrying cell within 10 000 (sensitivity ∼10−4) and is highly specific (Fueller et al17 ; supplemental Table 1 [available online at the Blood Web site]; supplemental Figure 1). False-positive results have not been obtained so far and the theoretical likelihood is very low (<10−10; supplemental Figure 2). The detection rates for ETV6-RUNX1+ and TCF3-PBX1+ by GIPFEL in previously tested, clinically known primary leukemia samples were 64% and 39%, respectively. Breakpoints outside the covered breakpoint region or complex genomic rearrangements were not detected.17
To detect the TCF3-PBX1 fusion, genomic DNA is digested with MfeI, ligated to obtain circular DNA, and then used as a template for semi-nested multiplexed real-time PCR (RT-PCR) to amplify the newly generated ligation joint. Amplification can occur only in case of a translocation, when DNA molecules from 2 chromosomes are joined. In case of a positive signal, primers are de-multiplexed and the product is checked by gel electrophoresis and Sanger sequencing for expected size and sequence. Chromosomal breakpoints of positive samples are PCR amplified and Sanger sequenced (supplemental Figure 3).
Here, we used GIPFEL for a retrospective screening of UCB samples from 340 healthy newborns for TCF3-PBX1 translocations. This study was approved by the Danish Data Protection Agency, the Danish Scientific Ethics Committee, and the Ethical Review Board of the University Hospital, Heinrich Heine University, Düsseldorf, Germany. Written informed consent was obtained from the parents. UCB samples from 340 healthy neonates were collected and processed within 24 hours from birth as described.17 Mononuclear cells were separated by Ficoll density centrifugation and stored in liquid nitrogen. GIPFEL screening was carried out after enrichment of B cells from cryopreserved UCB and isolation of genomic DNA as described previously.5,17
In 2 of 340 UCB samples from healthy newborns (N15 and N141), GIPFEL screening generated positive RT-PCR signals compatible with TCF3-PBX1 translocations. In both cases, the signal was validated by PCR amplification and Sanger sequencing of the ligation joints (Figure 1A-D; supplemental Figure 4). The results suggested that ∼0.6% of newborns harbor clonally expanded TCF3-PBX1+ B cells at levels detectable by GIPFEL.
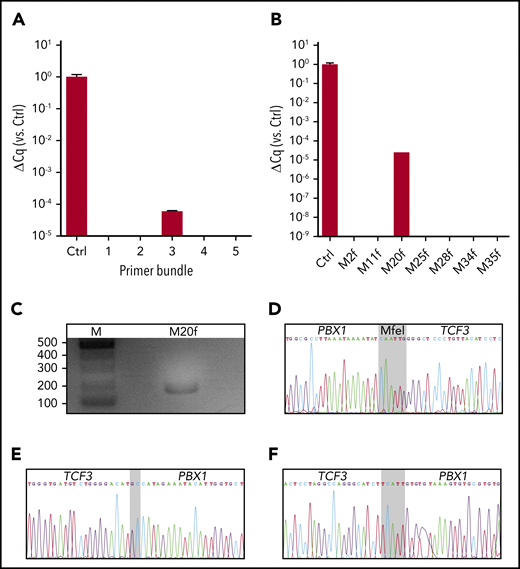
TCF3-PBX1+ GIPFEL results for samples N15 and N141. (A) Result of the first RT-PCR analysis of N15. The amplification of the 5 PBX1 primer bundles (1 through 5) was compared with the amplification of the internal PBX1 wild-type control (Ctrl). Primer bundle 3 leads to a detectable amplification. (B) Amplification plot of the second RT-PCR analysis of N15. The forward primers of bundle 3 were de-multiplexed, and primer PBX1-M20f was identified as the primer responsible for the amplification. (C) Agarose gel analysis of N15 after a PCR assay with PBX1-M20f and the reverse primer TCF3-M1r-n. The PCR data show the expected product of 158 bp. (D) Sanger sequencing result of UCB sample N15. The expected sequences flanking the MfeI ligation joint (gray) were identified, indicating the fusion of PBX1 segment M20 to TCF3 segment M1. (E) Sanger sequencing result of the chromosomal breakpoint of sample N15. Bases on a gray background indicate nontemplate bases. (F) Same as (E), but for sample N141. ΔCq, normalized quantification cycle, with the PBX1 wild-type control set as 1.
TCF3-PBX1+ GIPFEL results for samples N15 and N141. (A) Result of the first RT-PCR analysis of N15. The amplification of the 5 PBX1 primer bundles (1 through 5) was compared with the amplification of the internal PBX1 wild-type control (Ctrl). Primer bundle 3 leads to a detectable amplification. (B) Amplification plot of the second RT-PCR analysis of N15. The forward primers of bundle 3 were de-multiplexed, and primer PBX1-M20f was identified as the primer responsible for the amplification. (C) Agarose gel analysis of N15 after a PCR assay with PBX1-M20f and the reverse primer TCF3-M1r-n. The PCR data show the expected product of 158 bp. (D) Sanger sequencing result of UCB sample N15. The expected sequences flanking the MfeI ligation joint (gray) were identified, indicating the fusion of PBX1 segment M20 to TCF3 segment M1. (E) Sanger sequencing result of the chromosomal breakpoint of sample N15. Bases on a gray background indicate nontemplate bases. (F) Same as (E), but for sample N141. ΔCq, normalized quantification cycle, with the PBX1 wild-type control set as 1.
As in the case of the ETV6-RUNX1 fusion (5% ETV6-RUNX1+ for newborns compared with 0.01% ETV6-RUNX1+ leukemia rate), the frequency exceeds the corresponding TCF3-PBX1+ leukemia incidence (∼0.002%), and it is likely still underestimated because the detection rate of GIPFEL is less than 100% (about 39% for TCF3-PBX1+ fusions).17
We also used GIPFEL to estimate the frequencies at which these translocation-carrying B cells occurred. The frequencies of TCF3-PBX1+ B cells (N15, 6 × 10−5; N141, 1 × 10−3) were in a range similar to that determined for ETV6-RUNX1 (10−5 to 10−2) in our study and in other studies,4,5,18,19 and they indicate clonal expansion.
To identify the individual-specific breakpoints on a DNA level with bp resolution, we used Sanger sequencing. GIPFEL indicated that the maximum PCR product length for N15 and N141 would be 5139 bp and 3950 bp, respectively, when amplifying the breakpoint region by using a TCF3 primer in exon 16 and sample-specific reverse primers covering the potential PBX1 breakpoint region. Products of 2026 bp and 1390 bp were amplified for N15 and N141, respectively, and were sequenced by using the PCR primers. For N15, an additional set of primers lying closer to the breakpoint had to be used to identify the breakpoint (supplemental Table 2). By using this approach, we finally confirmed the GIPFEL results (Figure 1E-F). The breakpoint positions indicate that the fusion likely provides functional in-frame transcripts.
Because of anonymized sample processing, we could not track leukemia development in newborns of the analyzed cohort, and the impact of the detected fusions on leukemogenesis in these children remains undetermined. Larger cohorts with respective follow-up will be needed to clarify the translocation bearers’ risk of developing leukemia and identify factors that determine leukemia development.
To investigate whether breakpoints in healthy newborns differ from breakpoints in leukemia patients, we collected all available data on TCF3-PBX1 breakpoint sequences (supplemental Table 3).9,15,20 The TCF3 breakpoint for N141 lies in a known breakpoint cluster,21,22 and the TCF3 breakpoint for N15 lies in close proximity to this cluster (Figure 2). Breakpoints in PBX1 were more evenly distributed, even though all reported breakpoints lie in the 3′ half of the breakpoint cluster region. The breakpoint for N15 is the most distant 5′ breakpoint ever reported (Figure 2). Our data indicate that breakpoints in healthy individuals lie in similar regions; however, larger cohorts need to be analyzed.
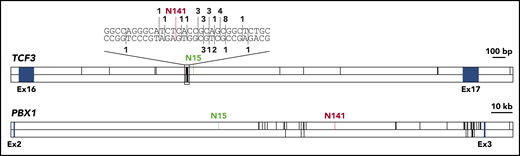
Distribution of TCF3-PBX1 breakpoints. The breakpoint cluster regions of TCF3 and PBX1 are shown. Blue boxes indicate exons. Known TCF3-PBX1 breakpoints are indicated by black vertical lines in the upper panel of each gene. Reciprocal PBX1-TCF3 breakpoints are depicted in the lower panel of each gene. For TCF3, a cluster region within the breakpoint cluster region is shown on bp resolution above the gene. Here, numbers indicate the number of reported breaks at this position. TCF3-PBX1 breaks are noted above the DNA strand, PBX1-TCF3 breaks below. The breaks reported in this study are shown in green (N15) and red (N141). Published breakpoints are taken from Wiemels et al,9 Paulsson et al,20 Fischer et al,15 and previously unpublished breakpoints (M.M., unpublished data, September 2001).
Distribution of TCF3-PBX1 breakpoints. The breakpoint cluster regions of TCF3 and PBX1 are shown. Blue boxes indicate exons. Known TCF3-PBX1 breakpoints are indicated by black vertical lines in the upper panel of each gene. Reciprocal PBX1-TCF3 breakpoints are depicted in the lower panel of each gene. For TCF3, a cluster region within the breakpoint cluster region is shown on bp resolution above the gene. Here, numbers indicate the number of reported breaks at this position. TCF3-PBX1 breaks are noted above the DNA strand, PBX1-TCF3 breaks below. The breaks reported in this study are shown in green (N15) and red (N141). Published breakpoints are taken from Wiemels et al,9 Paulsson et al,20 Fischer et al,15 and previously unpublished breakpoints (M.M., unpublished data, September 2001).
Our findings of the prenatal origin of TCF3-PBX1 are partially supported by the Wiemels et al study,9 which showed that in 2 of 15 patients, the TCF3-PBX1+ leukemia could be traced back to Guthrie cards. Our data do not rule out that the translocation can also occur postnatally. However, similar to ETV6-RUNX1+ ALL, this type of ALL is frequent in children but rarer in adults. It is therefore tempting to speculate that TCF3-PBX1+ preleukemic clones are prenatally formed and either convert to overt leukemia or, in most cases, die out later in life.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Sabine Hornhardt and Mathias Kessler (Federal Office for Radiation Protection, Oberschleissheim, Germany) for advice and support, Mel Greaves and Tony Ford (The Institute of Cancer Research, London, United Kingdom) for valuable comments, and Bianca Killing for excellent technical support.
This work was supported by the German Federal Office for Radiation Protection grants (36 14 S 30034 and 36 18 S 32275), by the Katharina Hardt Foundation, by intramural grants (2016-70, 2019-03) from the Research Commission of the Medical Faculty of the Heinrich Heine University (Düsseldorf), by the Danish Cancer Society, and by the Danish Childhood Cancer Foundation.
Authorship
Contribution: D.H. performed laboratory work, designed the research, analyzed data, and wrote the article; K.D. recruited the probands, analyzed clinical data, and performed laboratory work; M.M. provided clinical samples and analyzed clinical data; S.I. designed the research; K.S. designed and supervised the research; A.B. designed the research and wrote the article; U.F. designed the research, analyzed data, and wrote the article; and U.F. and A.B. were the principal investigators and take primary responsibility for the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ute Fischer, Department of Pediatric Oncology, Hematology, and Clinical Immunology, Center for Child and Adolescent Health, Heinrich Heine University, Moorenstr 5, 40225 Düsseldorf, Germany; e-mail: ute.fischer@med.uni-duesseldorf.de.

