Abstract
As essential components of hemoglobin, iron and heme play central roles in terminal erythropoiesis. The impairment of this process in iron/heme deficiency results in microcytic hypochromic anemia, the most prevalent anemia globally. Heme-regulated eIF2α kinase, also known as heme-regulated inhibitor (HRI), is a key heme-binding protein that senses intracellular heme concentrations to balance globin protein synthesis with the amount of heme available for hemoglobin production. HRI is activated during heme deficiency to phosphorylate eIF2α (eIF2αP), which simultaneously inhibits the translation of globin messenger RNAs (mRNAs) and selectively enhances the translation of activating transcription factor 4 (ATF4) mRNA to induce stress response genes. This coordinated translational regulation is a universal hallmark across the eIF2α kinase family under various stress conditions and is termed the integrated stress response (ISR). Inhibition of general protein synthesis by HRI-eIF2αP in erythroblasts is necessary to prevent proteotoxicity and maintain protein homeostasis in the cytoplasm and mitochondria. Additionally, the HRI–eIF2αP–ATF4 pathway represses mechanistic target of rapamycin complex 1 (mTORC1) signaling, specifically in the erythroid lineage as a feedback mechanism of erythropoietin-stimulated erythropoiesis during iron/heme deficiency. Furthermore, ATF4 target genes are most highly activated during iron deficiency to maintain mitochondrial function and redox homeostasis, as well as to enable erythroid differentiation. Thus, heme and translation regulate erythropoiesis through 2 key signaling pathways, ISR and mTORC1, which are coordinated by HRI to circumvent ineffective erythropoiesis (IE). HRI-ISR is also activated to reduce the severity of β-thalassemia intermedia in the Hbbth1/th1 murine model. Recently, HRI has been implicated in the regulation of human fetal hemoglobin production. Therefore, HRI-ISR has emerged as a potential therapeutic target for hemoglobinopathies.
Introduction
Iron deficiency (ID) anemia is a prevalent affliction worldwide and is a major contributor to the global burden of anemia.1 Most of the iron in the human body is bound in heme, where its inclusion in hemoglobin (Hb) accounts for as much as 70% of the total iron content.2 However, the molecular mechanisms by which iron and heme regulate erythropoiesis are still not fully understood. Heme-regulated eIF2α kinase, also known as heme-regulated inhibitor (HRI), is a unique heme-sensing protein with 2 heme-binding domains3-5 (Figure 1A); it is expressed predominantly in the erythroid lineage.6 In addition to serving as a prosthetic group for Hb, heme acts as a signaling molecule in gene expression by modulating HRI-mediated translation.7-9
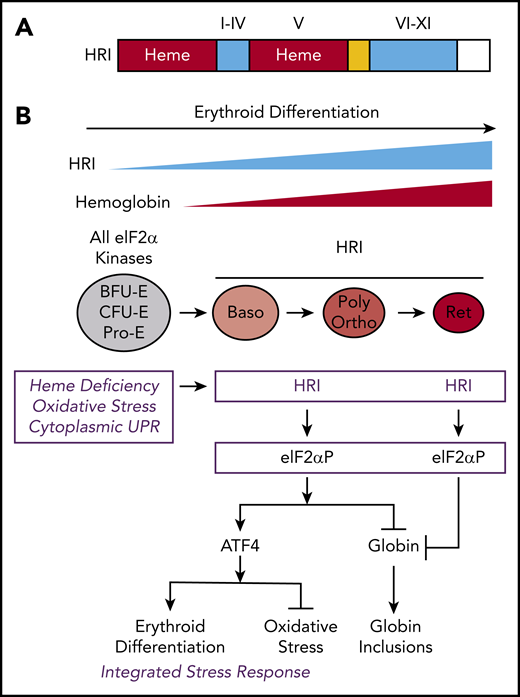
The protein structure and integrated stress response of HRI in erythropoiesis. (A) Heme and kinase domains of HRI. The 2 heme-binding domains in the N terminus and kinase insert (V) are marked and shaded in red, whereas the conserved kinase domains I-IV in the small lobe and VI-XI in the large lobe of protein kinase are shaded in blue. The N terminus, kinase insert, and C terminus are unique to HRI. (B) ISR of HRI during erythropoiesis. Expression of HRI and Hb intensify during terminal erythropoiesis from Baso, polychromatic (Poly), and orthochromatic (Ortho) erythroblasts to reticulocytes (Ret). Both arms of HRI-ISR, inhibition of globin synthesis and induction of ATF4 target gene expression, are operative in nucleated erythroblasts. In the enucleated reticulocytes, the role of HRI is limited to inhibit protein synthesis and, thus, prevent proteotoxicity of globin inclusions in heme deficiency.
The protein structure and integrated stress response of HRI in erythropoiesis. (A) Heme and kinase domains of HRI. The 2 heme-binding domains in the N terminus and kinase insert (V) are marked and shaded in red, whereas the conserved kinase domains I-IV in the small lobe and VI-XI in the large lobe of protein kinase are shaded in blue. The N terminus, kinase insert, and C terminus are unique to HRI. (B) ISR of HRI during erythropoiesis. Expression of HRI and Hb intensify during terminal erythropoiesis from Baso, polychromatic (Poly), and orthochromatic (Ortho) erythroblasts to reticulocytes (Ret). Both arms of HRI-ISR, inhibition of globin synthesis and induction of ATF4 target gene expression, are operative in nucleated erythroblasts. In the enucleated reticulocytes, the role of HRI is limited to inhibit protein synthesis and, thus, prevent proteotoxicity of globin inclusions in heme deficiency.
HRI is activated in heme deficiency and phosphorylates eIF2α, impairing further rounds of initiation, thereby inhibiting translation.8,10 During erythroid differentiation, HRI coordinates translation of globin messenger RNAs (mRNAs) with the availability of heme for the efficient production of large amounts of Hb in erythroid precursors.11 In HRI deficiency, excess globins synthesized during heme deficiency precipitate and cause proteotoxicity (Figure 1B).12,13 Beyond the regulation of globin translation, HRI reduces IE during ID and in β-thalassemia.12,13 IE occurring in Hri−/− mice during ID and in β-thalassemic mice is due primarily to the profound inhibition of erythroid differentiation starting at the basophilic (Baso) erythroblasts,14,15 which is also observed in Rb-deficient16,17 or Stat5a/5b-deficient18 mice. However, the underlying molecular mechanisms for the development of IE are not well understood.
Erythropoiesis under anemic stress is accelerated by elevated erythropoietin (Epo) levels in response to the hypoxia resulting from anemia.19 One of the Epo response pathways in erythroid precursors is phosphatidylinositol 3-kinase/AKT signaling and its downstream substrate mechanistic target of rapamycin complex 1 (mTORC1).20 mTORC1 signaling increases protein synthesis by phosphorylating eIF4E-binding proteins and ribosomal protein S6 kinase, which phosphorylates S6.21 Recently, it has been shown that the AKT/mTORC1/phosphorylated S6 axis is the major Epo signaling pathway in primary erythroid progenitors.22 Hyporesponsiveness to Epo therapy is commonly encountered in ID,23 indicating the presence of feedback mechanisms for homeostatic control of erythropoiesis by iron. The molecular mechanisms for such feedback are still largely unknown.
Regulation of reactive oxygen species (ROS) levels and oxidative stress is extremely important in erythropoiesis. Starting at Baso erythroblasts, erythroid precursors synthesize large amounts of Hb. Consequently, iron uptake for heme biosynthesis also increases, potentially generating ROS through the iron-catalyzed Fenton reaction.24 Oxidative stress occurring in β-thalassemia is 1 source of major complications in this disease and in other red cell disorders, such as sickle cell anemia (SCA). In addition to heme deficiency, HRI is activated by oxidative stress25,26 and denatured cytoplasmic proteins25,27 (Figure 1B), both of which occur in thalassemia.28 Indeed, HRI is required to reduce the phenotypic severity of the Hbbth1/th1 mouse model of β-thalassemia intermedia.13 The molecular basis of erythroid cell adaptation to oxidative stress is not fully understood. Furthermore, induction of fetal Hb (HbF) has been documented to ameliorate the symptoms of β-thalassemia and SCA.29-31 Recently, HRI has been implicated in regulating γ-globin gene expression by unknown mechanisms.32-34
Although transcriptional regulation during erythropoiesis has been studied extensively,35,36 much less is known about how translation may control this process.37,38 This review will focus on recent advancements in heme and HRI-mediated in vivo translation on protein homeostasis, oxidative stress, mitochondrial function, erythroid differentiation, and Epo signaling in primary murine erythroblasts. The emerging role of HRI in γ-globin gene expression will also be discussed.
Activation of HRI-ISR in primary erythroblasts
In addition to inhibiting translation of highly translated mRNAs, phosphorylate eIF2α (eIF2αP) selectively increases translation of certain poorly translated mRNAs for adaptation to stress (Figure 1B). This coordinated translational regulation has been termed the “integrated stress response” (ISR).39-41 Translational upregulation by eIF2αP requires upstream open reading frames (uORFs) in the 5' untranslated region (5′UTR) of these unique mRNAs, most notably ATF4 (activating transcription factor 4).42,43
In the erythroid lineage, HRI expression increases during terminal erythropoiesis44 (Figure 1B). Starting at Baso erythroblasts, HRI is the predominant eIF2α kinase responsible for >90% of eIF2α phosphorylation,44 and it is expressed at levels that are 2 orders of magnitude higher than the other 3 eIF2α kinases: PERK, GCN2, and PKR.45,46 These eIF2α kinases are expressed in distinct tissues to combat different stresses via ISR. PERK is activated by endoplasmic reticulum (ER) stress.47 PKR responds to viral infections,48 whereas GCN2 senses nutrient starvation.49
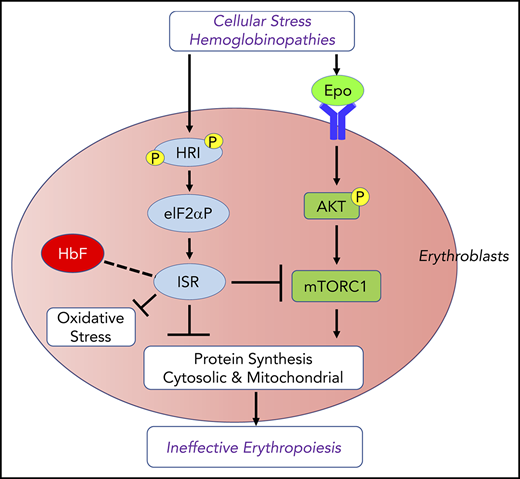
HRI activates the eIF2αP-ATF4 signaling pathway in primary erythroblasts to mitigate oxidative stress14,50 (Figure 1B). Hri−/− erythroblasts suffer from increased ROS and apoptosis upon acute oxidative stress induced by sodium arsenite. Induction of antioxidant genes upon acute oxidative stress in erythroblasts depends on HRI and ATF4. Impairment of the HRI-eIF2αP-ATF4 pathway generates red blood cells (RBCs) that are more sensitive to oxidative insult.14 During chronic ID in vivo, HRI is also necessary to reduce oxidative stress. ROS levels in RBCs and erythroid precursors are dramatically elevated during ID in Hri−/− mice but not in wild-type (Wt) mice.14,50 Thus, HRI-eIF2αP-ATF4 signaling provides a third signaling axis for combatting oxidative stress in addition to the Foxo3- and Nrf2-mediated pathways.51-53 The HRI-eIF2αP-ATF4 pathway may also cross talk with the Nrf2 pathway. ATF4 has been shown to interact with Nrf2 to regulate heme oxygenase 1 expression.54,55
Severe anemia and IE in mice defective in erythroid eIF2αP and Atf4
To better define the role of eIF2αP in mediating HRI stress signaling during erythropoiesis, an erythroid-specific eIF2α Ala51 knockin mouse model (eAA mice), which harbors a substitution of the phosphorylation site Ser51 with Ala, has been generated.50 eAA mice are also instrumental in determining whether eIF2α is the sole substrate of HRI and the mediator of enhanced translation of Atf4 mRNA in vivo. Despite activation of HRI, eIF2αP levels were greatly reduced in specifically Ter119+eAA erythroid cells compared with those from Wt mice. In iron-sufficient conditions (+Fe), eAA and Atf4−/− mice did not exhibit significant erythroid abnormalities. During ID, eAA and Atf4−/− mice were more anemic, with reduced RBC numbers and lower blood Hb levels than Wt and Hri−/− mice. eAA and Atf4−/− mice developed splenomegaly with elevated serum Epo levels in ID, similar to Hri−/− mice (Table 1). However, insoluble globin inclusions were visible in Hri−/− and eAA blood smears but not in Atf4−/− samples. Importantly, eAA mice developed macrocytic hyperchromic anemia in ID, similarly to Hri−/− mice.12 In contrast, Atf4−/− mice displayed microcytic hypochromic anemia similarly to Wt mice. Thus, eIF2αP, but not ATF4, is essential to reduce proteotoxicity from cytoplasmic unfolded heme-free globins and is required for the development of characteristic microcytic hypochromic anemia during ID. The similar erythroid phenotypes of eAA and Hri−/− mice demonstrate that the major physiological function of HRI in the erythroid lineage is mediated by eIF2αP.50
ATF4 and eIF2αP are necessary for erythroid differentiation and ROS reduction in ID
In Wt−Fe mice, ATF4 protein levels increased in lineage-depleted erythroid precursors from the spleen, but not from BM,50 in agreement with the spleen being the major organ for stress erythropoiesis. HRI and eIF2αP are necessary for ATF4 induction in vivo during ID.50
In iron sufficiency, differentiation was similar between mutant and Wt mice, with the exception of a mild erythroid expansion observed in the spleen of Hri−/− and eAA mice. In ID, eAA mice exhibited a similar inhibition of differentiation as did Hri−/− mice, starting at the Baso erythroblasts.50 Although most of the Atf4−/− mice died neonatally,56 the few surviving mice displayed normal steady-state erythropoiesis in iron sufficiency. During ID, half of the Atf4−/− mice exhibited splenic erythroid expansion with inhibition of differentiation similar to Hri−/− and eAA mice. Atf4−/− mice without splenic erythroid expansion exhibited an earlier inhibition in differentiation, with fewer erythroblasts and reticulocytes in the spleen and few Ter119+ cells in the BM.50 These findings demonstrate that late erythroblasts are the populations that undergo expansion in Hri−/−, eAA, and Atf4−/− mice during ID. Hri−/−, eAA, and Atf4−/− erythroid cells also exhibited significant increases in ROS levels in the BM, spleen, and blood, especially in late erythroblasts, reticulocytes, and RBCs, during ID.50
Repression of Epo-mTORC1 signaling by HRI-ISR in ID
mTORC1 signaling in blood cells was inhibited under ID, and mice with constitutively activated mTORC1 in the hematopoietic lineage exhibit macrocytic hyperchromic anemia with splenomegaly and IE,57 similar to the phenotypes of Hri−/− and eAA mice during ID. These observations support the notion that HRI-ISR signaling is necessary for repressing mTORC1 activity during ID (Figure 2). Indeed, HRI-ISR mutant mice displayed elevated mTORC1 activity during ID, specifically in the erythroid lineage.50
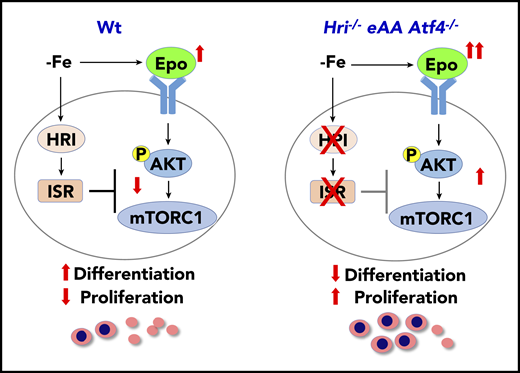
HRI-ISR represses Epo-mTORC1 signaling in iron-restricted erythropoiesis. In Wt mice, HRI is activated, phosphorylates eIF2α, and induces ISR during ID (−Fe). At the same time, the elevated serum Epo levels resulting from ID anemia induce AKT-mTORC1 signaling to increase protein synthesis and cell proliferation. HRI-ISR is necessary to repress mTORC1 activity and cell proliferation and, thus, promote terminal differentiation. In Hri−/−, eAA, and Atf4−/−mice, Epo-mTORC1 signaling remains active during ID as a result of defective HRI-ISR. This active Epo-mTORC1 signaling causes IE in ISR-defective mutant mice. Repression of mTORC1 by HRI-ISR serves as 1 feedback mechanism for terminating Epo signaling in ID. Reprinted from Zhang et al.50
HRI-ISR represses Epo-mTORC1 signaling in iron-restricted erythropoiesis. In Wt mice, HRI is activated, phosphorylates eIF2α, and induces ISR during ID (−Fe). At the same time, the elevated serum Epo levels resulting from ID anemia induce AKT-mTORC1 signaling to increase protein synthesis and cell proliferation. HRI-ISR is necessary to repress mTORC1 activity and cell proliferation and, thus, promote terminal differentiation. In Hri−/−, eAA, and Atf4−/−mice, Epo-mTORC1 signaling remains active during ID as a result of defective HRI-ISR. This active Epo-mTORC1 signaling causes IE in ISR-defective mutant mice. Repression of mTORC1 by HRI-ISR serves as 1 feedback mechanism for terminating Epo signaling in ID. Reprinted from Zhang et al.50
Treatment with rapamycin and INK128, mTORC1 inhibitors, greatly increased RBC numbers and Hb levels, as well as significantly reduced splenomegaly and Epo levels in Hri−/−−Fe mice. Additionally, rapamycin and INK128 promoted erythroid differentiation in the BM and spleens of Hri−/− mice, with a significant decreased percentage of early erythroblasts and increased percentage of orthochromatic erythroblasts, reticulocytes, and RBCs. Therefore, inhibition of mTORC1 signaling reduces IE of Hri−/− mice when iron is limiting. However, inhibition of mTORC1 did not significantly reduce globin inclusions or ROS in blood reticulocytes or RBCs of Hri−/− mice, demonstrating an essential and independent role for HRI-ISR in inhibiting globin mRNA translation and oxidative stress.50
Reversal of elevated HRI and mTORC1 signaling in ID by iron repletion
Dietary iron repletion (FeR) to iron-deficient Wt, Hri−/−, eAA, and Atf4−/− mice rapidly restored blood RBC numbers and Hb levels in Hri−/−, eAA, and Atf4−/− mice to levels similar to in Wt mice. Globin inclusions in reticulocytes of Hri−/− and eAA blood were no longer visible, and the RBC morphology of Atf4−/− was improved. Oxidative stress in the reticulocytes of iron-deficient Hri−/−, eAA, and Atf4−/− blood samples was also relieved upon FeR with normal ROS levels. Most importantly, HRI hyperphosphorylation and protein levels in the spleen of eAA and Atf4−/− mice were greatly reduced upon FeR. Furthermore, mTORC1 activity was significantly reduced upon FeR in erythroid cells of Hri−/−, eAA, and Atf4−/− mice concomitantly with reduced splenomegaly and serum Epo levels, as well as increased erythroid differentiation.50
As summarized in Figure 2, HRI coordinates 2 key pathways of translation, eIF2αP and mTORC1 signaling, for effective iron-restricted erythropoiesis. Induction of ATF4 expression by HRI-eIF2αP is required in ID to repress Epo-stimulated mTORC1 signaling and IE, and it serves as a feedback mechanism for homeostatic control of erythropoiesis by iron and heme. eIF2αP is also necessary in ID for adaptation to microcytic hypochromic anemia and to inhibit globin translation, thereby reducing proteotoxicity. This role of HRI-ISR in reducing unfolded cytoplasmic globin and promoting differentiation of erythroid cells during ID parallels the function of PERK-ISR in reducing proinsulin translation and maintaining pancreatic β-cells in the differentiated state,58 highlighting tissue-specific mechanisms of activating eIF2α kinases in regulating proteostasis of highly and uniquely expressed proteins in specialized erythroid and pancreatic tissues.
Preferential translation of Atf4 mRNA in vivo by HRI during terminal erythropoiesis
Although the specific role of HRI in the translational regulation of globin and Atf4 mRNAs in erythroid cells has been appreciated,9,14,50 an understanding of the global impact of HRI-mediated translational regulation on erythropoiesis has only recently emerged through ribosome profiling (Ribo-seq) studies of Baso erythroblasts from Wt+Fe, Wt−Fe, Hri−/−+Fe, and Hri−/−−Fe fetal livers (FLs).46 Ribo-seq is a powerful tool to interrogate translation genome wide in an unbiased manner59 (Figure 3).
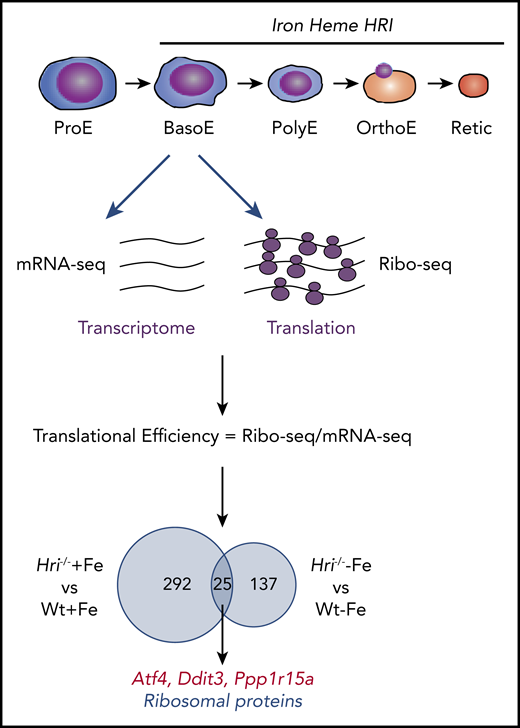
Genome-wide in vivo translation assessment by ribosome profiling in iron and HRI deficiencies. Iron, heme, and HRI are critical for terminal erythropoiesis from Baso erythroblasts (BasoE) to reticulocytes (Retic). BasoE were sorted by flow cytometry using CD71 and Ter119 surface markers from embryonic day 14.5 FLs of Wt and Hri−/− embryos under +Fe and −Fe conditions. Actively translated mRNAs are occupied by ribosomes, which protect fragments of mRNAs from RNase digestion. cDNA libraries from ribosome-protected fragments and polyA+ mRNAs were subjected to DNA sequencing, followed by mapping to mouse genome mm10.46 Translational efficiency (TE) of each mRNA was calculated as the ratio of Ribo-seq reads/mRNA-seq reads. Venn diagrams of numbers of differential translated mRNAs between Wt and Hri−/− BasoEs under +Fe and −Fe are shown. Under both +Fe and −Fe conditions, TE of Atf4 mRNA is most highly enhanced in Wt BasoEs compared with Hri−/− cells, whereas translation of ribosomal protein mRNAs is most highly enhanced in Hri−/− BasoEs compared with Wt cells. Adapted from Zhang et al with permission.46
Genome-wide in vivo translation assessment by ribosome profiling in iron and HRI deficiencies. Iron, heme, and HRI are critical for terminal erythropoiesis from Baso erythroblasts (BasoE) to reticulocytes (Retic). BasoE were sorted by flow cytometry using CD71 and Ter119 surface markers from embryonic day 14.5 FLs of Wt and Hri−/− embryos under +Fe and −Fe conditions. Actively translated mRNAs are occupied by ribosomes, which protect fragments of mRNAs from RNase digestion. cDNA libraries from ribosome-protected fragments and polyA+ mRNAs were subjected to DNA sequencing, followed by mapping to mouse genome mm10.46 Translational efficiency (TE) of each mRNA was calculated as the ratio of Ribo-seq reads/mRNA-seq reads. Venn diagrams of numbers of differential translated mRNAs between Wt and Hri−/− BasoEs under +Fe and −Fe are shown. Under both +Fe and −Fe conditions, TE of Atf4 mRNA is most highly enhanced in Wt BasoEs compared with Hri−/− cells, whereas translation of ribosomal protein mRNAs is most highly enhanced in Hri−/− BasoEs compared with Wt cells. Adapted from Zhang et al with permission.46
Beginning at Baso erythroblasts, erythropoiesis is finely regulated by iron and heme levels60,61 (Figure 3). More than 300 mRNAs were significantly differentially translated in HRI deficiency under +Fe conditions, supporting the role of HRI in normal fetal erythropoiesis. Twenty-five differentially translated mRNAs were common under +Fe and −Fe conditions, including the well-characterized ISR mRNAs Atf4, Ppp1r15a, and Ddit3 (Figure 3). Each of these mRNAs contains uORFs in their 5′UTR, and their translation is upregulated by eIF2αP in cell lines under ER stress or amino acid starvation.41 Ribosome occupancies at uORFs of these mRNAs were also observed in vivo in primary erythroblasts in an HRI-dependent manner.46
Atf4 mRNA was the most differentially translated mRNA between Wt and Hri−/− cells during ID.46 Atf4 mRNA has 2 well-characterized uORFs in its 5′UTR, including uORF1, which encodes 3 aa and is translated regardless of stress and eIF2αP levels.41 Atf4 uORF1 had exceptionally high ribosome occupancy in vivo. However, uORF2 and the canonical open reading frame of Atf4 were poorly translated in HRI deficiency under +Fe and −Fe conditions. Thus, Atf4 mRNA is positioned for HRI-mediated translation in developing erythroblasts. Notably, Hri and Atf4 mRNAs are abundantly expressed in Baso erythroblasts at levels similar to those of Gata1 and Fog1 (Zfpm1), master erythroid transcription factors.46 Interestingly, Atf4 mRNA is most highly expressed in Ter119+CD71+ erythroblasts among the 16 differentiation stages of murine bone marrow hematopoietic cells,62 underscoring the significance of HRI-enhanced translation of Atf4 mRNA in terminal erythropoiesis.
Maintenance of ribosome homeostasis and mitochondrial function by HRI in ID
In HRI- and iron-deficient states, translation of mRNAs encoding ribosomes and mitochondrial proteins is most significantly upregulated (Figure 4A). Translation initiation and elongation, as well as mTORC1 signaling, were the most highly elevated pathways in HRI deficiency, including increased formation of the 43S translation initiation complex,46 consistent with such a function of HRI-eIF2αP.8 Notably, 56 ribosomal protein mRNAs were more highly translated in iron and HRI deficiencies. Among them, translation of the 18 ribosomal protein mRNAs lacking 5′ terminal oligopyrimidine motifs was regulated by HRI but not mTORC1.46
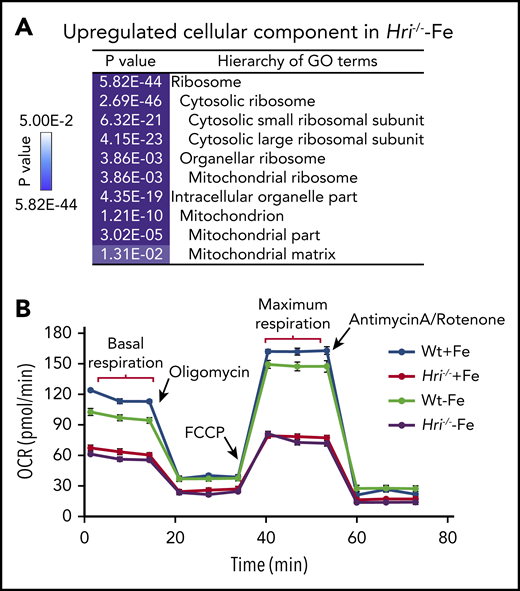
HRI inhibits translation of nuclear encoded mRNAs of mitochondrial proteins and maintains mitochondrial respiration in ID. (A) In addition to inhibiting cytosolic ribosomal protein synthesis, HRI is necessary to inhibit translation of mRNAs encoding mitochondrial ribosomal proteins, oxidative phosphorylation proteins, and matrix proteins during ID. (B) In the absence of HRI, the increased translation of these mRNAs in ID results in increased mitochondrial protein synthesis, which impairs mitochondrial respiration, likely as a result of proteotoxicity. Adapted from Zhang et al with permission.46
HRI inhibits translation of nuclear encoded mRNAs of mitochondrial proteins and maintains mitochondrial respiration in ID. (A) In addition to inhibiting cytosolic ribosomal protein synthesis, HRI is necessary to inhibit translation of mRNAs encoding mitochondrial ribosomal proteins, oxidative phosphorylation proteins, and matrix proteins during ID. (B) In the absence of HRI, the increased translation of these mRNAs in ID results in increased mitochondrial protein synthesis, which impairs mitochondrial respiration, likely as a result of proteotoxicity. Adapted from Zhang et al with permission.46
Translation of 163 mRNAs of nuclear-encoded mitochondrial proteins was upregulated in iron and HRI deficiencies, including transcripts for mitochondrial ribosomal proteins, components necessary for oxidative phosphorylation (encoding for complexes I through V), and matrix proteins.46 Interestingly, this set of differentially translated mitochondrial protein mRNAs in HRI deficiency was distinct from those mitochondrial proteins upregulated by mTORC1/phosphorylating eIF4E-binding proteins–mediated translation.63 This difference may be due, in part, to different cellular context, murine primary erythroblasts46 vs human breast cancer cell line,63 and the conditions for modulating mTORC1 activities. Nonetheless, increased translational efficiency (TE) of mitochondrial proteins was accompanied by increased mitochondrial protein synthesis in iron and HRI deficiencies. mTORC1 pathway contributed to ∼50% of mitochondrial protein synthesis in Wt and Hri−/− primary erythroblasts.46 Mitochondrial protein synthesis was still elevated in Hri−/− primary erythroblasts upon inhibition of mTORC1 activity.46 Therefore, HRI-eIF2αP contributes directly to regulation of mitochondrial protein synthesis during ID, in addition to its role in repressing mTORC1 activity.
Although Wt erythroid cells were able to maintain a normal oxygen consumption rate in ID, Hri−/− erythroid cells displayed decreased basal and maximal respiration under +Fe and −Fe conditions46 (Figure 4B). This impaired mitochondrial respiration in Hri−/− erythroid cells was not due to the reduced mitochondrial mass or DNA content in these cells. Thus, HRI is necessary to maintain mitochondrial respiration by inhibiting mitochondrial protein synthesis. Mitochondria are the energy powerhouses of the cell and are also necessary for amino acid metabolism, nucleotide production, and heme and iron-sulfur cluster biosynthesis.64 Translational regulation of mitochondrial biogenesis by mTORC1 is particularly important for erythropoiesis because of the high demand of heme for Hb production and oxidative stress.65 Interestingly, the mRNAs upregulated translationally at proerythroblasts65 during normal erythropoiesis are also distinct from those regulated by HRI at Baso erythroblasts in ID.46 It is important to note that hyperactive and hypoactive mTORC1 activities result in impaired erythropoiesis and anemia.57,65 Thus, a fine balance of appropriate mTORC1 activity is critical during erythropoiesis, and HRI may serve as a regulator opposing mTORC1 in regulating mitochondrial function, especially under stress conditions.
In mitochondrial disorders, mitochondrial unfolded protein response (UPRmt) is activated to coordinate with nuclear transcription to enable restoration of mitochondrial function.64 Interestingly, ISR is also activated in mitochondrial dysfunction and mediates UPRmt.66-68 However, the eIF2α kinase responsible for UPRmt remains unknown. Observations that Hri−/− erythroid cells have impaired mitochondrial function and have no significant increased expression of mitochondrial chaperones suggest that HRI may be the eIF2α kinase responsible for activation of UPRmt and, thereby, contribute to the proper maintenance of mitochondrial function of erythroid cells to ID.46
Cytoplasmic unfolded protein response in iron and HRI deficiencies
The most upregulated process in the transcriptome during iron and HRI deficiencies is protein folding and refolding, which includes many cytoplasmic chaperone genes from the Hsp70, Hsp90, Hsp40, and Hsp110 families46 (Figure 5). ER and mitochondrial chaperones were not significantly increased. Increased expression of Hsp70 and Hsp90 was observed in erythroid cells of Hri−/− and eAA mice in ID.12,50 Cytoplasmic chaperones were not induced in the presence of HRI during ID, further supporting the essential and primary role of HRI in inhibiting protein synthesis to coordinate protein homeostasis with available iron and heme concentrations to mitigate proteotoxicity. However, activation of the cytoplasmic unfolded protein response alone is not sufficient to compensate for the loss of HRI. Indeed, Hri−/− FL erythroid precursors accumulated significant amounts of protein inclusions during terminal differentiation, resulting in cell death with fragments of cell debris46 (Figure 5). In contrast, Atf4−/− FL erythroid cells did not suffer from proteotoxicity as expected, because of the presence of functional HRI-eIF2αP to inhibit translation. However, differentiation of Atf4−/− erythroid precursors was inhibited with the accumulation of myeloid cells.46,50 These findings further support that enhanced translation of Atf4 mRNA is required for erythroid differentiation.
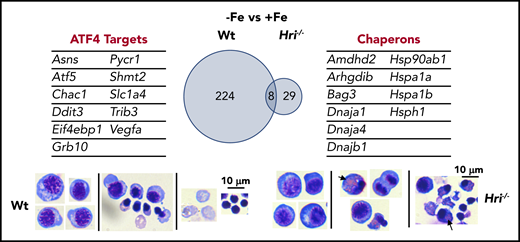
HRI-ISR prevents cytoplasmic unfolded protein response and enables terminal erythropoiesis. Differentially expressed mRNAs between +Fe and −Fe conditions of Wt and Hri−/− BasoEs are shown. Only 37 mRNAs are differentially expressed in Hri−/− BasoEs during ID, in contrast to 232 mRNAs that are differentially expressed in Wt BasoEs. Importantly, ATF4 target genes are most highly activated, specifically in Wt BasoEs, during ID to mitigate oxidative stress and to enable terminal erythropoiesis. Thus, HRI is necessary for transcriptome adaptation to ID. The majority of upregulated mRNAs are cytoplasmic chaperons in Hri−/− BasoEs during ID as a response to denatured unfolded globin synthesized in excess of heme. However, increased chaperone expression alone in the absence of HRI is not sufficient to overcome the proteotoxicity. Hri−/− FL erythroid progenitors suffer severe proteotoxicity of aggregated protein inclusions during terminal erythroid differentiation from 30 hours to 42 hours when Hb synthesis is intensified. Lower panels, C57/BL6 stain. Adapted from Zhang et al with permission.46
HRI-ISR prevents cytoplasmic unfolded protein response and enables terminal erythropoiesis. Differentially expressed mRNAs between +Fe and −Fe conditions of Wt and Hri−/− BasoEs are shown. Only 37 mRNAs are differentially expressed in Hri−/− BasoEs during ID, in contrast to 232 mRNAs that are differentially expressed in Wt BasoEs. Importantly, ATF4 target genes are most highly activated, specifically in Wt BasoEs, during ID to mitigate oxidative stress and to enable terminal erythropoiesis. Thus, HRI is necessary for transcriptome adaptation to ID. The majority of upregulated mRNAs are cytoplasmic chaperons in Hri−/− BasoEs during ID as a response to denatured unfolded globin synthesized in excess of heme. However, increased chaperone expression alone in the absence of HRI is not sufficient to overcome the proteotoxicity. Hri−/− FL erythroid progenitors suffer severe proteotoxicity of aggregated protein inclusions during terminal erythroid differentiation from 30 hours to 42 hours when Hb synthesis is intensified. Lower panels, C57/BL6 stain. Adapted from Zhang et al with permission.46
HRI-ATF4 is necessary for transcriptome adaptation to ID
The global transcriptional impact of ID and the role of HRI in mediating the cellular response to ID were revealed by gene-expression profiling studies. Significantly more genes are differentially expressed between Wt−Fe and Wt+Fe Baso erythroblasts than between Hri−/−−Fe and Hri−/−+Fe cells (232 vs 37; Figure 5), demonstrating the near-absolute requirement for HRI in regulating the transcriptional response to ID.46
In ID, ATF4 target genes10 are highly induced in erythroblasts and dependent on HRI. These upregulated ISR genes are also activated upon ER stress39 and are involved in serine-glycine biosynthesis, 1 carbon metabolism, proline-glutamine synthesis, and glutathione metabolism. Furthermore, the expression levels of these genes are lower in HRI deficiency even under +Fe conditions, suggesting that HRI fine-tunes the ISR during iron replete erythropoiesis and, thus, has an important role, even under normal conditions. Interestingly, all of these ATF4 target genes are expressed at higher levels during normal primitive erythropoiesis compared with definitive erythropoiesis,45 suggesting that HRI-ISR is also operative and important in primitive erythropoiesis.
HRI-ATF4–mediated GRB10 induction as a feedback mechanism of Epo-stimulated erythropoiesis in ID
One of the most highly induced ATF4 target genes, growth factor receptor-bound protein 10 (Grb10), has been reported to be part of a feedback mechanism to inhibit growth factor–mediated mTORC1 signaling, such as insulin69 and stem cell factor.70 Grb10 has also been shown to be a GATA2 target gene and play a role in the transition from burst-forming unit-erythroid cells to colony-forming unit-erythroid cells.71
A reduction in Grb10 expression by short hairpin RNAs in FL erythroid progenitors increased cell numbers of differentiating erythroblasts and inhibited terminal erythroid differentiation with the accumulation of polychromatic erythroblasts and a decrease in orthochromatic erythroblasts,46 recapitulating the hallmarks of IE observed in Hri−/− mice in ID.12,14,50 Thus, ATF4-induced Grb10 expression by HRI-ISR serves as a feedback mechanism for Epo signaling during ID in terminal erythropoiesis during which GATA2 expression is low.
In summary, studies of genome wide in vivo translation and transcription uncover the pleiotropic functions of heme and HRI-regulated translational control that involves several separate and interacting pathways for the adaptation to systemic ID (Figure 6). Activation of HRI in ID elicits 3 distinct pathways of translation: first and foremost, inhibition of protein synthesis to avoid accumulation of unfolded proteins and maintain protein homeostasis; induction of Atf4 mRNA translation to increase gene expression for mitochondrial unfolded protein response, redox homeostasis, and metabolic reprogramming to maintain mitochondrial respiration and enable erythroid differentiation; and repression of Epo-mTORC1 signaling by induction of GRB10 to inhibit cell proliferation and enable erythroid differentiation.
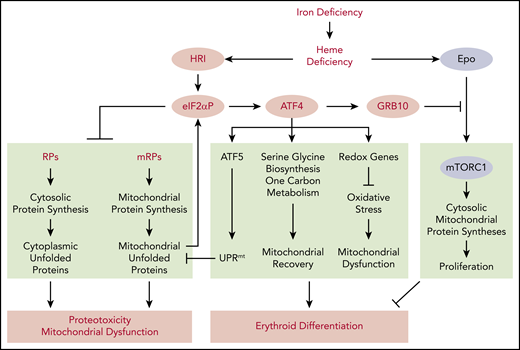
Global impact of HRI-mediated gene expression in vivo in primary erythroblasts during ID. Global impact of HRI-mediated in vivo translation in primary BasoE was investigated by Ribo-seq and mRNA-seq, as illustrated in Figure 3. An in vivo heme deficiency mouse model via diet-induced systemic ID was used.12 HRI is activated by heme deficiency and phosphorylates eIF2α. First, eIF2αP inhibits general protein synthesis in cytoplasm and mitochondria through inhibiting the translation of ribosomal protein mRNAs in the cytosolic and mitochondrial cellular compartments. In the absence of HRI, continued protein synthesis results in the accumulation of cytoplasmic and mitochondrial unfolded proteins leading to proteotoxicity and mitochondrial dysfunction. Second, eIF2αP selectively enhances the translation of Atf4 mRNA. ATF4 then induces gene expression of 3 pathways in activating UPRmt, reprogramming mitochondrial metabolism and reducing oxidative stress, all of which enable adaptation to ID and erythroid differentiation. Last, HRI-ISR suppresses mTORC1 signaling, which is activated by elevated Epo levels in ID, via ATF4-induced GRB10 expression. Overall, global genome-wide gene expression assessment of primary erythroblasts in vivo reveals that HRI-ISR contributes most significantly to adaptation to iron-restricted erythropoiesis. Reprinted from Zhang et al with permission.46
Global impact of HRI-mediated gene expression in vivo in primary erythroblasts during ID. Global impact of HRI-mediated in vivo translation in primary BasoE was investigated by Ribo-seq and mRNA-seq, as illustrated in Figure 3. An in vivo heme deficiency mouse model via diet-induced systemic ID was used.12 HRI is activated by heme deficiency and phosphorylates eIF2α. First, eIF2αP inhibits general protein synthesis in cytoplasm and mitochondria through inhibiting the translation of ribosomal protein mRNAs in the cytosolic and mitochondrial cellular compartments. In the absence of HRI, continued protein synthesis results in the accumulation of cytoplasmic and mitochondrial unfolded proteins leading to proteotoxicity and mitochondrial dysfunction. Second, eIF2αP selectively enhances the translation of Atf4 mRNA. ATF4 then induces gene expression of 3 pathways in activating UPRmt, reprogramming mitochondrial metabolism and reducing oxidative stress, all of which enable adaptation to ID and erythroid differentiation. Last, HRI-ISR suppresses mTORC1 signaling, which is activated by elevated Epo levels in ID, via ATF4-induced GRB10 expression. Overall, global genome-wide gene expression assessment of primary erythroblasts in vivo reveals that HRI-ISR contributes most significantly to adaptation to iron-restricted erythropoiesis. Reprinted from Zhang et al with permission.46
HRI-ISR is necessary to reduce the severity of β-thalassemia
Accumulation of unpaired α-globin, oxidative stress, and IE are hallmarks of β-thalassemia,28 and all of these biological processes can be regulated by HRI-ISR, as described above. HRI is necessary to mitigate the phenotypic severity of Hbbth1/th1 β-thalassemic mice. Hri−/−Hbbth1/th1 mice were embryonically lethal, whereas Hri+/−Hbbth1/th1 mice developed more severe symptoms, including proteotoxicity of α-globin inclusions, IE, and iron overload.13 HRI is activated and induces Atf4 mRNA translation in mouse β-thalassemic erythroid precursors.13,14 Salubrinal, a selective inhibitor of eIF2αP dephosphorylation,72 increases eIF2αP and reduces globin inclusions in β-thalassemic reticulocytes. Furthermore, salubrinal enhances the eIF2αP signaling pathway in β-thalassemic erythroid precursors by increasing ATF4 and CHOP proteins.14 These findings provide the foundation for exploiting the HRI-ISR signaling pathway for treatment of thalassemia (Figure 7).
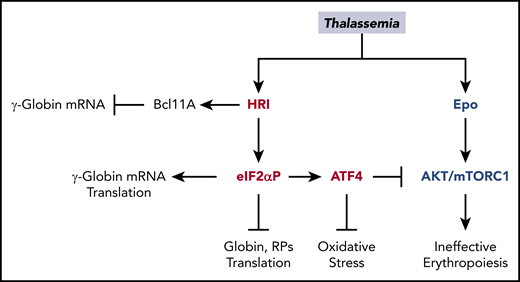
Multiple roles of HRI in attenuating the severity of thalassemia phenotypes. In β-thalassemia, HRI is activated to inhibit excess α-globin synthesis and, thus, reduces proteotoxicity. HRI-induced translation of ATF4 mRNA activates downstream gene expression to mitigate oxidative stress. Furthermore, the HRI-ATF4 axis represses Epo-mTORC signaling to reduce IE. In human erythropoiesis, HRI is implicated in fetal γ-globin expression in an opposing manner. On one hand, HRI was reported to increase translation of γ-globin mRNA32,33 ; however, on the other hand, it was shown to inhibit transcription of γ-globin gene by increasing Bcl11A mRNA.34
Multiple roles of HRI in attenuating the severity of thalassemia phenotypes. In β-thalassemia, HRI is activated to inhibit excess α-globin synthesis and, thus, reduces proteotoxicity. HRI-induced translation of ATF4 mRNA activates downstream gene expression to mitigate oxidative stress. Furthermore, the HRI-ATF4 axis represses Epo-mTORC signaling to reduce IE. In human erythropoiesis, HRI is implicated in fetal γ-globin expression in an opposing manner. On one hand, HRI was reported to increase translation of γ-globin mRNA32,33 ; however, on the other hand, it was shown to inhibit transcription of γ-globin gene by increasing Bcl11A mRNA.34
Role of HRI in human fetal γ-globin expression
In human erythropoiesis, salubrinal was shown to increase eIF2αP levels and HbF production, with a concomitant decrease in HbA in differentiating CD34+ cells, by increasing translation of γ-globin mRNA while inhibiting general translation32,33 (Figure 7). Importantly, this eIF2αP-mediated pathway works synergistically with 2 clinical therapeutics, azacitidine and hydroxyurea, to induce higher levels of HbF than can be achieved by a single agent alone.32 Although polysome profiling studies revealed an increased TE of γ-globin mRNA relative to β-globin mRNA, the underlying mechanism remains to be elucidated.33
On the other hand, HRI was recently identified as a potential repressor for HbF production by a domain-focused CRISPR genome-editing screen in HUDEP2 cells.34 HRI depletion increased the expression of HbF and γ-globin mRNA concomitantly with the diminished expression of Bcl11a mRNA, a repressor of γ-globin transcription31,73,74 (Figure 7). More importantly, depletion of HRI in primary CD34+ cells from SCA patients increased γ-globin mRNA and HbF to levels sufficient to reduce sickling under low oxygen tension. Depletion of HRI also worked synergistically with the pharmacological reagent pomalidomide, which induced HbF, in part by reducing BCL11A.75 Interestingly, this HbF-repressor action of HRI appears to be unique to humans and was not observed in murine erythroid cells.34
Expression of Bcl11a, embryonic (Hbb-y), and fetal-like (Hbb-bh1) murine β-globins from Ribo-seq studies of primary murine Baso erythroblasts46 is shown in Table 2. At the translational level, TE of Bcl11a mRNA was not significantly altered in HRI deficiency and/or ID. Interestingly, TE of Hbb-y and Hbb-bh1 mRNAs was significantly decreased in HRI deficiency, especially under normal +Fe, in agreement with the role of HRI in enhancing human γ-globin mRNA translation.32,33 At the transcriptome level, no significant change in embryonic and fetal β-globins was observed, consistent with the finding of Grevet et al that the HbF-repressor action of HRI appears to be unique to humans.34 It is noteworthy that Bcl11a mRNA was not altered in HRI deficiency alone, but it was significantly decreased in iron and HRI deficiencies. This difference in Bcl11a mRNA expression by HRI between human and mouse systems is intriguing and awaiting further investigation.
As summarized in Figure 7, the evidence for protective functions of HRI-ISR in β-thalassemia is exceedingly strong, especially in the murine model.13,14 In addition, HRI-ISR is activated in human cancer cell lines and primary multiple myeloma cells,76,77 and it has emerged as a molecular target of anticancer agents.78,79 Therefore, HRI-ISR is conserved between mice and humans. The recent enthusiasm in inducing HbF via inhibition of HRI-ISR34 will need to take into the consideration the pleiotropic roles of HRI during terminal erythropoiesis and, in particular, under the stress conditions of hemoglobinopathies (Figure 7).
Concluding remarks
Recent studies have demonstrated the impact of heme and HRI-eIF2αP–mediated translational repression of globin mRNAs and translational activation of Atf4 mRNA during erythropoiesis, especially under iron/heme deficiency and β-thalassemia. The repression of globin mRNA translation by HRI reduces proteotoxicity, as well as permits the expression of ATF4 protein, which plays a pivotal role in terminal erythropoiesis by maintaining oxidative homeostasis and mitochondrial functions. Furthermore, HRI-ATF4–mediated gene expression represses mTORC1 signaling and provides a feedback mechanism to attenuate Epo-mTORC1–stimulated IE in ID anemia. Inhibition of mTORC1 also improves anemia and promotes erythroid differentiation of Foxo3−/−, β-thalassemic, and SCA mice.80,81 Thus, elevated Epo-mTORC1 signaling may be a common phenomenon in IE. Furthermore, improved pathology in thalassemic mice upon iron restriction82-84 is likely to be mediated, in part, by the repression of mTORC1 signaling and globin mRNA translation by HRI. Combination therapy using iron/heme restriction and mTORC1 inhibitors may work synergistically to improve hemoglobinopathies of thalassemia and SCA. The recent discovery that ribosomal protein synthesis is regulated by HRI46 also suggests a potential role for HRI in the pathology of ribosomopathies, such as Diamond-Blackfan anemia.38
Although much is known about the signaling of Epo in stimulating erythropoiesis, less is understood about the termination of Epo signaling. GRB10,46 TFR2,85 and Scribble86 are 3 recently discovered proteins that enable feedback loops in Epo signaling during ID. Future studies to elucidate the molecular mechanisms underlying hyporesponsiveness to Epo will be invaluable to our understanding of the regulation of erythropoiesis in disease states of ID anemia, hemoglobinopathies, and anemia of inflammation.
The newly identified functions of HRI-ATF4 in mitochondrial 1 carbon amino acid metabolism and mitochondrial respiration open a new direction for investigations that focus on the roles of metabolites and metabolism in the regulation of erythropoiesis. Recently, serine and glycine production has been shown to be important for terminal erythroid erythropoiesis.87 Glutamine-dependent nucleotide biosynthesis is also required for Epo-stimulated erythropoiesis of hematopoietic stem cells.88 Recent findings regarding HRI’s role in fetal γ-globin expression warrant further investigation of HRI biology in human erythropoiesis, especially in relation to globin switching, which differs in the murine system, as well as the extent to which this regulatory process can be decoupled from the pleiotropic roles of HRI in other aspects of erythropoiesis.
Acknowledgments
The authors thank Vijay G. Sankaran (Harvard Medical School and Boston Children’s Hospital) for critical reading of the manuscript and helpful comments.
Research in the authors’ laboratory was supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK087984 (J.-J.C.).
Authorship
Contribution: J-J.C. wrote the manuscript and designed figures, and S.Z. edited the manuscript and designed and prepared figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for S.Z. is Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan 250012, People’s Republic of China.
Correspondence: Jane-Jane Chen, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, 77 Mass Ave, Room E25-421A, Cambridge, MA 02139; e-mail: j-jchen@mit.edu.