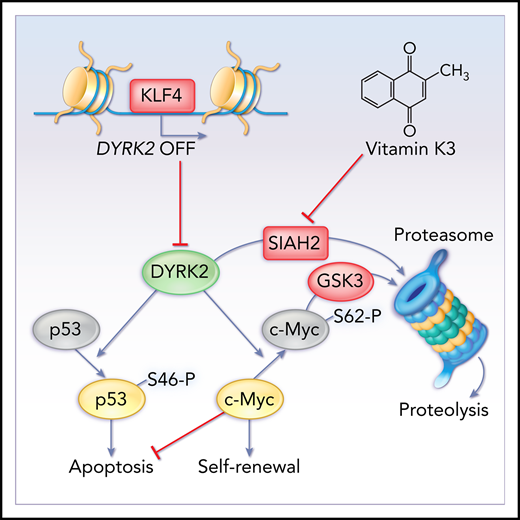
Key Points
Loss of KLF4 impairs self-renewal and survival in CML stem/progenitor cells through derepression of the DYRK2 gene.
Stabilization of DYRK2 protein inhibits survival and self-renewal in leukemia stem/progenitor cells via c-Myc depletion and p53 activation.

Abstract
Leukemia stem cells are a rare population with a primitive progenitor phenotype that can initiate, sustain, and recapitulate leukemia through a poorly understood mechanism of self-renewal. Here, we report that Krüppel-like factor 4 (KLF4) promotes disease progression in a murine model of chronic myeloid leukemia (CML)-like myeloproliferative neoplasia by repressing an inhibitory mechanism of preservation in leukemia stem/progenitor cells with leukemia-initiating capacity. Deletion of the Klf4 gene severely abrogated the maintenance of BCR-ABL1(p210)–induced CML by impairing survival and self-renewal in BCR-ABL1+ CD150+ lineage-negative Sca-1+ c-Kit+ leukemic cells. Mechanistically, KLF4 repressed the Dyrk2 gene in leukemic stem/progenitor cells; thus, loss of KLF4 resulted in elevated levels of dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (DYRK2), which were associated with inhibition of survival and self-renewal via depletion of c-Myc protein and p53 activation. In addition to transcriptional regulation, stabilization of DYRK2 protein by inhibiting ubiquitin E3 ligase SIAH2 with vitamin K3 promoted apoptosis and abrogated self-renewal in murine and human CML stem/progenitor cells. Altogether, our results suggest that DYRK2 is a molecular checkpoint controlling p53- and c-Myc–mediated regulation of survival and self-renewal in CML cells with leukemic-initiating capacity that can be targeted with small molecules.
Introduction
Leukemia stem cells (LSCs) generated by the transformation of normal hematopoietic stem/progenitor cells are elusive targets for therapy that can initiate and sustain leukemia owing to their unique capacity to regenerate themselves during self-renewing cell division while continuously feeding the neoplasm.1-6 Therefore, a better understanding of the mechanisms of self-renewal specific to LSCs is essential to overcome the inability of current chemotherapeutic drugs to safely eliminate this population and to prevent relapses.
Chronic myeloid leukemia (CML) is a type of stem cell leukemia that originates through the constitutive activation of BCR-ABL1 kinase, which is generated by the chromosomal translocation t(9;22) known as the Philadelphia chromosome.2,7-9 This myeloid neoplasm is normally diagnosed in the initial chronic phase; however, if left untreated it can progress through an accelerated phase to a lethal blast crisis driven by reprogrammed myeloid progenitor cells. CML can be successfully managed using tyrosine kinase inhibitors (TKIs) that suppress BCR-ABL1 activity, and patients remain in remission as long as they adhere to lifelong treatment because of the survival of LSCs that develop BCR-ABL1–independent mechanisms of self-renewal and survival.10 However, discontinuation trials have shown success and safety in a select group of patients, with at least half achieving treatment-free remission after the cessation of drug therapy, although some patients experience significant adverse events, and treatment discontinuation requires patient consent and knowledge of risks and benefits.2,11-14 These findings suggest that a cure may not be possible with TKIs alone, and new breakthroughs in CML therapy (primarily the identification of novel mechanisms of leukemic self-renewal) are urgently needed to eradicate disease with LSC-specific drugs. Treatment-free remissions will also reduce the health care costs associated with treatment and the emotional and financial burdens in a growing population of CML patients in lifelong therapy.2,3,12,15-18
The transcription factor Krüppel-like factor 4 (KLF4) has essential roles in the control of self-renewal in embryonic stem cells, reprogramming somatic cells into pluripotent stem cells, and carcinogenesis.19-25 Potential antitumor activity has been ascribed to KLF4 in B-cell non-Hodgkin and Hodgkin lymphomas, multiple myeloma, and acute myeloid leukemia.26-29 Furthermore, we recently reported that KLF4 prevents the expansion of leukemia-initiating cells by repressing the kinase MAP2K7 in T-cell acute lymphoblastic leukemia.30 Here, we report that conditional deletion of the Klf4 gene impairs the maintenance of leukemia in a model of CML-like myeloproliferative neoplasia caused by numerical and functional losses of leukemia stem/progenitor cells. Gene expression, promoter activity, and chromatin immunoprecipitation analyses revealed that KLF4 represses expression of the dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (DYRK2), which is involved in protein stability, cell cycle control, and apoptosis31-34 and is also known for promoting proteasomal degradation of c-Myc and c-Jun in HeLa cells and apoptosis in osteosarcoma and colorectal cancer cell lines.35,36 In our model, loss of KLF4 resulted in impaired survival and abrogation of self-renewal via p53 activation and c-Myc depletion in leukemic stem/progenitor cells. Finally, we showed that in vivo inhibition of SIAH2 with vitamin K3 (VK3) induces apoptosis and abrogates self-renewal in murine and human CML stem/progenitor cells by augmenting DYRK2 protein levels. In summary, our study provides insights into a novel mechanism of self-renewal specific to CML cells with self-renewal and leukemia-initiating capacity and reveals DYRK2 as a critical checkpoint in the control of LSC maintenance and a potential target with a dual function of abrogating self-renewal and survival for the development of LSC-targeted drugs to treat CML.
Methods
Mouse model of CML-like neoplasia
To induce CML-like disease in mice, we collected bone marrow cells from the femurs and tibias of untreated control (Klf4fl/flVav-iCre−; referred to as Klf4fl/fl) mice or KLF4-deficient (Klf4fl/flVav-iCre+; referred to as Klf4Δ/Δ) mice (8- to 10-week-old females) and purified hematopoietic stem cells (HSCs) as lineage-negative Sca-1+ c-Kit+ (LSK) cells positive for CD50 (LSK CD150+ cells). LSK CD150+ cells were plated on RetroNectin-coated plates and transduced with BCR-ABL1 retrovirus in the presence of Polybrene (8 μg/mL) by spinoculation (60 minutes at 456 relative centrifugal force).37 Retroviruses were generated by cotransfection of the MSCV-IRES-GFP construct carrying the BCR-ABL1 (p210) complementary DNA and pEco packaging plasmid in 293T cells. After transduction (15%-22% GFP+), the cells were cultured for 2 to 3 days in the presence of stem cell factor (100 ng/mL), interleukin-3 (6 ng/mL), and interleukin-6 (10 ng/mL) in X-VIVO 15 medium, and GFP+ cells were purified by cell sorting. For transplantation, 800 GFP+ LSK CD150+ cells mixed with 400 000 radioprotective bone marrow cells were injected IV into recipient mice that were previously irradiated with 950 rad. The development of leukemia was monitored in peripheral blood by flow cytometric detection of Gr1, CD11b, F4/80, and B220 within leukemic cells (GFP+). For secondary transplantation, GFP+ LSK cells were isolated from CML mice at 14 days posttransplantation (dpt), immediately before attrition of Klf4Δ/Δ leukemia stem/progenitor cells, and transplanted into lethally irradiated C57BL/6J mice (1500 cells per mouse with 400 000 radioprotective bone marrow cells). The mice were monitored for the expansion of leukemia cells in peripheral blood, overall survival, and tissue distribution of leukemia cells. For other experiments, the number and phenotype of transplanted cells are indicated in the corresponding figure.
Statistical analysis
All sample sizes (n values) reported in each figure legend correspond to independent biological replicates. Experiments were performed without blinding and with no exclusion of samples. Experimental results with >8 values were confirmed to follow a normal distribution using the D'Agostino-Pearson normality test, and no significant difference in the variance between groups was detected using the F test (GraphPad Prism). An unpaired 2-tailed Student t test was used for statistical analysis. The survival of leukemic mice was visualized using Kaplan-Meier curves, and statistical significance was calculated using the log-rank test (GraphPad Prism). P values were determined using GraphPad software. Results with a P value < .05 were considered statistically significant.
Additional methods can be found in supplemental Material and Methods (available on the Blood Web site).
Results
KLF4 supports BCR-ABL1–induced CML-like leukemia by maintaining the pool of leukemic stem/progenitor cells
The role of KLF4 in CML is largely unknown, despite an enrichment of KLF4 transcripts in LSCs from CML patients (Figure 1A) and murine CML LSCs (supplemental Figure 1). To induce a CML-like myeloproliferative disease in mice, we used Cre-recombinase driven by the Vav promoter for gene deletion in hematopoietic cells (supplemental Figure 1).38 LSK CD150+ cells, a cell population that is highly enriched in HSCs,39 were purified by cell sorting of bone marrow cells from Klf4Δ/Δ and Klf4fl/fl mice. LSK CD150+ cells were then transformed by retroviral transduction using the MSCV-IRES-GFP retrovirus carrying BCR-ABL1(p210) and GFP, sorted for GFP, and transplanted in small numbers (to mimic chronic disease development) into cytoablated wild-type mice with radioprotective bone marrow (Figure 1B).37 In contrast to mice in the wild-type group, a significantly lower number of mice in the group transplanted with BCR-ABL1+Klf4Δ/Δ cells displayed expansion of myeloid leukemia cells (CD11b+ Gr1+) (Figure 1C; supplemental Figure 1). Interestingly, the Klf4Δ/Δ CML group showed slower disease progression and lower penetrance caused by myeloid leukemia (1/10) or B-cell leukemia (2/10), whereas the control group developed leukemia (7/10) more rapidly (Figure 1D-E). Immunophenotypic analysis showed the expansion of myeloid cells (CD11b+ Gr1+) in the majority of mice from both groups at 20 dpt, with a minor percentage of leukemic cells with a monocytic (CD11b+ Gr1−) or B-cell (B220+) phenotype, whereas most wild-type mice succumbed to CML-like neoplasia (Figure 1E). In the Klf4Δ/Δ group, 1 mouse died from myeloid leukemia, whereas the other 2 succumbed to B-cell leukemia, despite an initial expansion of CD11b+ Gr1+ cells (Figure 1E; supplemental Figure 2). To determine whether KLF4 is required for the maintenance of preestablished leukemia stem/progenitor cells, we induced deletion of the Klf4 gene after engraftment of BCR-ABL1+ cells using the tamoxifen-inducible ROSA-CreERT2 model (Figure 1F). Interestingly, a similar improvement in survival was observed after administration of tamoxifen to mice transplanted with BCR-ABL1–transduced Klf4fl/flROSA-CreERT2+ (induced Δ/Δ, iΔ/Δ) HSCs compared with that in control mice also treated with tamoxifen (Figure 1G), with most mice dying from myeloid leukemia in both groups, supporting a therapeutic benefit of inhibiting KLF4 downstream targets in patients with established disease. To identify the cause of unstable leukemia in the absence of KLF4, we first examined the frequency of leukemia stem/progenitor cells in bone marrow at 18 dpt. Klf4Δ/Δ-leukemic mice showed a significant reduction in GFP+ LSK cells and GFP+ LSK cells lacking expression of Flt3 (LSK Flt3− cells) in bone marrow (Figure 1H-I) and the spleen (Figure 1I). Notably, no significant differences were detected in these populations when gating on nonleukemic (GFP−) bone marrow cells in the same animals (data not shown).
![Loss of KLF4 impairs the maintenance of BCR-ABL1–induced CML-like disease. (A) KLF4 expression in normal and leukemic stem (CD34+ CD38− ALDHhi) and progenitor (CD34+ CD38+) cells (n = 5, mean ± standard deviation [SD]) was analyzed using published data (GSE43754). (B) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4Δ/Δ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted with radioprotective bone marrow (BM) into wild-type recipient mice to induce CML-like myeloproliferative neoplasia. (C) The expansion of myeloid leukemia cells (GFP+ CD11b+) in peripheral blood (fl/fl, n = 10; Δ/Δ, n = 10). (D) Kaplan-Meier analysis of survival in 2 representative experiments. (E) Immunophenotype of leukemic cells at 20 dpt and moribund mice. (F) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4fl/flRosa-CreER+ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted into wild-type recipient mice to induce gene deletion at 7 dpt. (G) Survival of leukemic mice after tamoxifen (Tam) injection in both groups (fl/fl, n = 7; iΔ/Δ, n = 4). Immunophenotype of leukemia in diseased mice is also shown (right panel). (H) Representative flow cytometric analysis of CML LSCs, defined as GFP+ Lin− Sca-1+ c-Kit+ (GFP+ LSK) cells and GFP+ LSK Flt3− cells, in the bone marrow of Klf4fl/fl (fl/fl) or Klf4fl/flVav-iCre+ (Δ/Δ) leukemic mice at 18 dpt (n = 5). (I) Enumeration of GFP+ LSK and GFP+ LSK Flt3− cells in the bone marrow (BM) and spleen of Klf4fl/fl or Klf4Δ/Δ CML mice (n = 5, mean ± SD). The data shown are representative of 3 independent experiments. *P < .05, **P < .01, ****P < .0001, 2-tailed Student t test. BMT, bone marrow transplantation; HPC, hematopoietic progenitor cells; iΔ/Δ, inducible Δ/Δ; LPC, leukemia progenitor cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2018875922/3/m_blood875922f1.png?Expires=1769470553&Signature=fownj7bYHAAA5Jovsvs~gMpUj~uYuWR3o2PcSS8t5ZQHs0swOXfV3mBxIz5aXQXAo0sZZxX7WZMcG3JtZ1s3RqRrW3UGObZsUEJvr5~Z63FpBBcpL9twOjIUjIe-h7wxz7pHzg3LlHOD6oVbQgpp7nAAvwV9g~rou76wpMV6EXrXA2IYuzxb5A0Amxf-0lwcDMDfThZ0CJFAGYVB7C9YznDWer7h0zDjAtsL6BhCMR2HsR1-8SX~x7HcXaUvNlE5Dpxx3D9nNDnhUR~6OmTjGaNXe~RNQ6iSrLTaHSfM3EzCJYVeISZ19vMZ60HUtSlU8LtMyGCI7dFPLb-3sR5LpQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Loss of KLF4 impairs the maintenance of BCR-ABL1–induced CML-like disease. (A) KLF4 expression in normal and leukemic stem (CD34+ CD38− ALDHhi) and progenitor (CD34+ CD38+) cells (n = 5, mean ± standard deviation [SD]) was analyzed using published data (GSE43754). (B) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4Δ/Δ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted with radioprotective bone marrow (BM) into wild-type recipient mice to induce CML-like myeloproliferative neoplasia. (C) The expansion of myeloid leukemia cells (GFP+ CD11b+) in peripheral blood (fl/fl, n = 10; Δ/Δ, n = 10). (D) Kaplan-Meier analysis of survival in 2 representative experiments. (E) Immunophenotype of leukemic cells at 20 dpt and moribund mice. (F) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4fl/flRosa-CreER+ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted into wild-type recipient mice to induce gene deletion at 7 dpt. (G) Survival of leukemic mice after tamoxifen (Tam) injection in both groups (fl/fl, n = 7; iΔ/Δ, n = 4). Immunophenotype of leukemia in diseased mice is also shown (right panel). (H) Representative flow cytometric analysis of CML LSCs, defined as GFP+ Lin− Sca-1+ c-Kit+ (GFP+ LSK) cells and GFP+ LSK Flt3− cells, in the bone marrow of Klf4fl/fl (fl/fl) or Klf4fl/flVav-iCre+ (Δ/Δ) leukemic mice at 18 dpt (n = 5). (I) Enumeration of GFP+ LSK and GFP+ LSK Flt3− cells in the bone marrow (BM) and spleen of Klf4fl/fl or Klf4Δ/Δ CML mice (n = 5, mean ± SD). The data shown are representative of 3 independent experiments. *P < .05, **P < .01, ****P < .0001, 2-tailed Student t test. BMT, bone marrow transplantation; HPC, hematopoietic progenitor cells; iΔ/Δ, inducible Δ/Δ; LPC, leukemia progenitor cells.
Loss of KLF4 impairs the maintenance of BCR-ABL1–induced CML-like disease. (A) KLF4 expression in normal and leukemic stem (CD34+ CD38− ALDHhi) and progenitor (CD34+ CD38+) cells (n = 5, mean ± standard deviation [SD]) was analyzed using published data (GSE43754). (B) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4Δ/Δ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted with radioprotective bone marrow (BM) into wild-type recipient mice to induce CML-like myeloproliferative neoplasia. (C) The expansion of myeloid leukemia cells (GFP+ CD11b+) in peripheral blood (fl/fl, n = 10; Δ/Δ, n = 10). (D) Kaplan-Meier analysis of survival in 2 representative experiments. (E) Immunophenotype of leukemic cells at 20 dpt and moribund mice. (F) LSK CD150+ cells from Klf4fl/fl (fl/fl) or Klf4fl/flRosa-CreER+ (Δ/Δ) mice were transduced with retrovirus containing BCR-ABL1-p210 and GFP and transplanted into wild-type recipient mice to induce gene deletion at 7 dpt. (G) Survival of leukemic mice after tamoxifen (Tam) injection in both groups (fl/fl, n = 7; iΔ/Δ, n = 4). Immunophenotype of leukemia in diseased mice is also shown (right panel). (H) Representative flow cytometric analysis of CML LSCs, defined as GFP+ Lin− Sca-1+ c-Kit+ (GFP+ LSK) cells and GFP+ LSK Flt3− cells, in the bone marrow of Klf4fl/fl (fl/fl) or Klf4fl/flVav-iCre+ (Δ/Δ) leukemic mice at 18 dpt (n = 5). (I) Enumeration of GFP+ LSK and GFP+ LSK Flt3− cells in the bone marrow (BM) and spleen of Klf4fl/fl or Klf4Δ/Δ CML mice (n = 5, mean ± SD). The data shown are representative of 3 independent experiments. *P < .05, **P < .01, ****P < .0001, 2-tailed Student t test. BMT, bone marrow transplantation; HPC, hematopoietic progenitor cells; iΔ/Δ, inducible Δ/Δ; LPC, leukemia progenitor cells.
KLF4 protects leukemia stem/progenitor cells from apoptosis and loss of self-renewal
The loss of leukemic stem/progenitor cells in the absence of KLF4 could be caused by alterations in cell division and/or survival. Analysis of DNA content revealed a small increase in the 2n DNA G0/G1 phase of Klf4Δ/Δ GFP+ LSK cells (Figure 2A), whereas Ki67/7-aminoactinomycin D analysis showed an increased proportion in the G0 phase (supplemental Figure 3). Next, we cultured purified GFP+ LSK cells in vitro to reveal their intrinsic survival capacity and observed a significant increase in Klf4Δ/Δ GFP+ LSK cells positive for annexin V under these conditions (Figure 2B). Consistent with this increase in cell death, we detected cleaved PARP by immunoblot analysis, primarily in Klf4Δ/Δ GFP+ LSK cells (Figure 2C). Next, we transplanted a 1:1 mixture of GFP+ LSK cells from wild-type (CD45.1.2) and Klf4Δ/Δ (CD45.2) leukemic mice to evaluate the role of KLF4 in the same recipient (Figure 2D). Remarkably, Klf4Δ/Δ leukemia cells were lost at 24 dpt, despite a similar engraftment at 15 dpt (Figure 2E), suggesting normal homing and cell-intrinsic attrition of leukemic stem/progenitor cells in the absence of KLF4. Transplantation of whole bone marrow cells from mice with primary leukemia induced death in all secondary recipient mice from CML-like disease using wild-type, but not Klf4Δ/Δ, leukemic bone marrow (supplemental Figure 4). This finding suggests a defective self-renewal capacity because development and maintenance of leukemia in secondary recipient mice rely on the ability of leukemia stem/progenitor cells to initiate and drive disease through self-renewing divisions. To further confirm defective self-renewal in Klf4Δ/Δ leukemia stem/progenitor cells and to evaluate functional defects via a cell-to-cell comparison during secondary transplantation, we purified GFP+ LSK cells from mice with primary CML-like disease 2 weeks posttransplantation (Figure 2F). All mice transplanted with the same number of Klf4Δ/Δ GFP+ LSK cells remained free of leukemia, despite an initial engraftment comparable to control mice (supplemental Figure 5), in sharp contrast to control mice that rapidly died of CML-like disease with 100% penetrance (Figure 2F; supplemental Figure 5). Further supporting a functional stem cell defect, compared with control leukemic stem/progenitor cells, Klf4Δ/Δ GFP+ LSK Flt3− leukemic cells purified from mice with primary myeloid leukemia were unable to generate colonies in methylcellulose cultures (Figure 2G). Finally, Klf4Δ/Δ GFP+ LSK CD150+ leukemic cells failed to serially replate in methylcellulose cultures, a surrogate assay of self-renewal, despite showing similar survival compared with control cells (Figure 2H). In summary, the inability of primitive progenitor leukemic cells to recapitulate leukemia and serially generate colonies in methylcellulose strongly supports a model in which KLF4 regulates self-renewal and survival in leukemia stem/progenitor cells that feed myeloid neoplasms.
![KLF4 promotes survival and self-renewal in leukemia stem/progenitor cells. (A) Cell cycle analysis of GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice was performed by flow cytometric detection of DNA content at 15 dpt. Statistical analysis of phases of the cell cycle (fl/fl, n = 5; Δ/Δ, n = 4) is also shown (right panel; data are mean ± standard deviation [SD]). (B) Cell death was determined by flow cytometric detection of annexin V in purified GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice 15 dpt after incubation for 24 hours (n = 4, mean ± SD). (C) Immunoblot analysis of PARP cleavage in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. (D) Transplantation of a 1:1 mixture of wild-type (WT; CD45.1+.2+) and Klf4Δ/Δ (CD45.2+) GFP+ LSK cells into lethally irradiated B6.SJL (CD45.1+) mice. (E) Flow cytometric analysis of donor-derived cells in peripheral blood at 15 dpt and 24 dpt, as described in (E) (n = 5, mean ± SD). (F) GFP+ LSK cells purified from Klf4fl/fl or Klf4Δ/Δ CML mice were transplanted into cytoablated mice to evaluate self-renewal. Kaplan-Meier survival curves of CML mice (fl/fl, n = 7; Δ/Δ, n = 3). (G) GFP+ LSK Flt3− cells were plated in methylcellulose cultures. (H) GFP+ LSK CD150+ cells were serially replated in methylcellulose to assess colony formation and annexin V staining in each plating (fl/fl, n = 5; Δ/Δ, n = 5, mean ± SD). Data are representative of 2 or 3 independent experiments. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test; P < .01, log-rank test. PI, propidium iodide.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2018875922/3/m_blood875922f2.png?Expires=1769470553&Signature=CPXNzS~aCTY58JdZhk-vdQ-8EDRr-LPSQPn3zm5gpOXexV6CfSTQrOg0IPyTCSPc4d-fE8E-LN~voa27zfRYF2w3YjJ7yQRpHCIUoRxyBIk4dc~R~rfWkeT64YwIp1I7P5nIRk71--i8pRdBudVk2xJ4pSGAp7rqcA1WrckCno8InvxuDUjok9GBgJlH0dKlITJkgv2ISyMzljp3WuSbo5HgQqoaGtCUADmEp~lqYs~5EJ1SXIVxgUY9ZrSPLH43N~o~tgVTJKCouvxaLKkDSX1son2fL0TbSzPFtSfaDOGeTgwJvpxihhKWVAE1HuaXE6BLpbTpPCIMyZVCImBhBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
KLF4 promotes survival and self-renewal in leukemia stem/progenitor cells. (A) Cell cycle analysis of GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice was performed by flow cytometric detection of DNA content at 15 dpt. Statistical analysis of phases of the cell cycle (fl/fl, n = 5; Δ/Δ, n = 4) is also shown (right panel; data are mean ± standard deviation [SD]). (B) Cell death was determined by flow cytometric detection of annexin V in purified GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice 15 dpt after incubation for 24 hours (n = 4, mean ± SD). (C) Immunoblot analysis of PARP cleavage in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. (D) Transplantation of a 1:1 mixture of wild-type (WT; CD45.1+.2+) and Klf4Δ/Δ (CD45.2+) GFP+ LSK cells into lethally irradiated B6.SJL (CD45.1+) mice. (E) Flow cytometric analysis of donor-derived cells in peripheral blood at 15 dpt and 24 dpt, as described in (E) (n = 5, mean ± SD). (F) GFP+ LSK cells purified from Klf4fl/fl or Klf4Δ/Δ CML mice were transplanted into cytoablated mice to evaluate self-renewal. Kaplan-Meier survival curves of CML mice (fl/fl, n = 7; Δ/Δ, n = 3). (G) GFP+ LSK Flt3− cells were plated in methylcellulose cultures. (H) GFP+ LSK CD150+ cells were serially replated in methylcellulose to assess colony formation and annexin V staining in each plating (fl/fl, n = 5; Δ/Δ, n = 5, mean ± SD). Data are representative of 2 or 3 independent experiments. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test; P < .01, log-rank test. PI, propidium iodide.
KLF4 promotes survival and self-renewal in leukemia stem/progenitor cells. (A) Cell cycle analysis of GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice was performed by flow cytometric detection of DNA content at 15 dpt. Statistical analysis of phases of the cell cycle (fl/fl, n = 5; Δ/Δ, n = 4) is also shown (right panel; data are mean ± standard deviation [SD]). (B) Cell death was determined by flow cytometric detection of annexin V in purified GFP+ LSK cells from Klf4fl/fl and Klf4Δ/Δ CML mice 15 dpt after incubation for 24 hours (n = 4, mean ± SD). (C) Immunoblot analysis of PARP cleavage in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. (D) Transplantation of a 1:1 mixture of wild-type (WT; CD45.1+.2+) and Klf4Δ/Δ (CD45.2+) GFP+ LSK cells into lethally irradiated B6.SJL (CD45.1+) mice. (E) Flow cytometric analysis of donor-derived cells in peripheral blood at 15 dpt and 24 dpt, as described in (E) (n = 5, mean ± SD). (F) GFP+ LSK cells purified from Klf4fl/fl or Klf4Δ/Δ CML mice were transplanted into cytoablated mice to evaluate self-renewal. Kaplan-Meier survival curves of CML mice (fl/fl, n = 7; Δ/Δ, n = 3). (G) GFP+ LSK Flt3− cells were plated in methylcellulose cultures. (H) GFP+ LSK CD150+ cells were serially replated in methylcellulose to assess colony formation and annexin V staining in each plating (fl/fl, n = 5; Δ/Δ, n = 5, mean ± SD). Data are representative of 2 or 3 independent experiments. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test; P < .01, log-rank test. PI, propidium iodide.
Identification of the kinase DYRK2 as a transcriptional target of KLF4 in leukemic stem/progenitor cells
To identify downstream targets of KLF4 responsible for the regulation of self-renewal and survival in leukemic stem/progenitor cells, we performed global gene expression analysis using GFP+ LSK cells purified by cell sorting from wild-type and Klf4Δ/Δ CML mice 2 weeks after transplantation. Bioinformatics analysis of biological triplicates for each group revealed genes with potential functions as novel regulators of self-renewal that were significantly deregulated in Klf4Δ/Δ leukemic cells, with cell growth and proliferation as 2 of the most altered cellular functions (Figure 3A-B) and deregulated expression of genes expressed in stem cells (supplemental Figure 6). We decided to further study DYRK2 because it was 1 of the most upregulated genes in Klf4Δ/Δ GFP+ LSK cells that provided the possibility of pharmacological modulation (Figure 3A). Immunoblot analysis showed increased DYRK2 protein in purified Klf4Δ/Δ GFP+ LSK cells (leukemic stem/progenitor cells) that was not observed in normal hematopoietic stem/progenitor cells (Figure 3C). To determine whether there is direct regulation, we performed chromatin immunoprecipitation coupled with polymerase chain reaction (ChIP-PCR) in lineage-negative bone marrow cells from CML mice and observed binding of endogenous KLF4 protein to the Dyrk2 promoter (−155 to +23) but not to upstream (−1032 to −793) and exon 2 (+7081 to +7917) sequences (Figure 3D). In addition, a luciferase promoter assay showed that KLF4 reduced Dyrk2-luciferase activity (Figure 3E). Therefore, increased expression of DYRK2 in Klf4Δ/Δ GFP+ LSK cells and the binding of KLF4 to the endogenous Dyrk2 promoter indicated that KLF4 inhibits Dyrk2 gene expression.
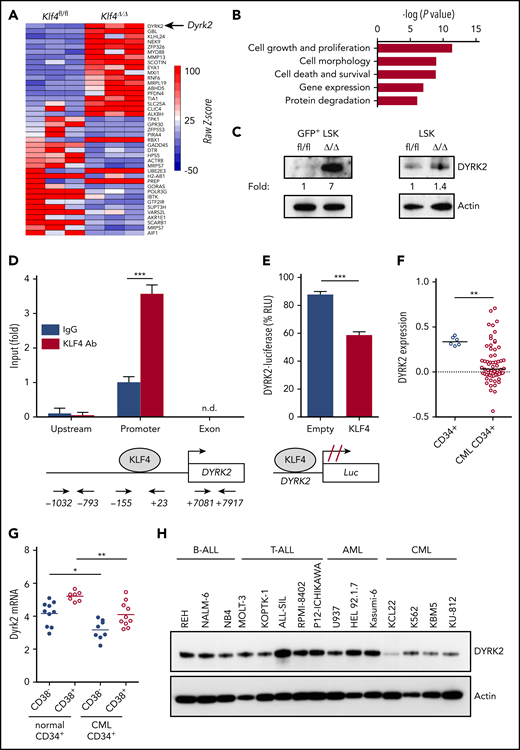
The kinase DYRK2 is repressed by KLF4 in leukemia stem/progenitor cells. (A) Gene expression heat map showing the most deregulated genes comparing GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. (B) Gene ontology analysis of deregulated cellular pathways using Ingenuity Pathway Analysis. (C) Immunoblot analysis of DYRK2 protein expression in purified GFP+ LSK cells (leukemia stem/progenitor cells) from the bone marrow of CML mice and LSK cells (hematopoietic stem/progenitor cells). Densitometry values were used to estimate the fold increase compared with wild-type using values normalized to actin. (D) Analysis of KLF4 occupancy at the DYRK2 promoter in lineage-negative bone marrow cells from CML mice by ChIP-PCR analysis. Genomic DNA was immunoprecipitated with anti-KLF4 antibody (KLF4 Ab) or IgG control and amplified with primers spanning the DYRK2 promoter, upstream, and exon sequences. (E) Promoter assay using a KLF4 expression plasmid and Dyrk2-luciferase reporter construct in the 293T cell line. (F) Analysis of DYRK2 messenger RNA levels in CD34+ cells from healthy individuals and CML patients in the chronic phase using a published data set (GSE4170). (G) DYRK2 qPCR in CD34+ CD38− (stem) and CD34+ CD38+ (progenitor) cells purified from CML patients and healthy donors. (H) Immunoblot analysis of DYRK2 expression in a panel of lymphoid and myeloid leukemia cell lines. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test (D-E), Mann-Whitney U test (F-G). n.d., not detected.
The kinase DYRK2 is repressed by KLF4 in leukemia stem/progenitor cells. (A) Gene expression heat map showing the most deregulated genes comparing GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. (B) Gene ontology analysis of deregulated cellular pathways using Ingenuity Pathway Analysis. (C) Immunoblot analysis of DYRK2 protein expression in purified GFP+ LSK cells (leukemia stem/progenitor cells) from the bone marrow of CML mice and LSK cells (hematopoietic stem/progenitor cells). Densitometry values were used to estimate the fold increase compared with wild-type using values normalized to actin. (D) Analysis of KLF4 occupancy at the DYRK2 promoter in lineage-negative bone marrow cells from CML mice by ChIP-PCR analysis. Genomic DNA was immunoprecipitated with anti-KLF4 antibody (KLF4 Ab) or IgG control and amplified with primers spanning the DYRK2 promoter, upstream, and exon sequences. (E) Promoter assay using a KLF4 expression plasmid and Dyrk2-luciferase reporter construct in the 293T cell line. (F) Analysis of DYRK2 messenger RNA levels in CD34+ cells from healthy individuals and CML patients in the chronic phase using a published data set (GSE4170). (G) DYRK2 qPCR in CD34+ CD38− (stem) and CD34+ CD38+ (progenitor) cells purified from CML patients and healthy donors. (H) Immunoblot analysis of DYRK2 expression in a panel of lymphoid and myeloid leukemia cell lines. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test (D-E), Mann-Whitney U test (F-G). n.d., not detected.
Consistent with an inhibitory role in CML GFP+ LSK cells, data mining of an available data set revealed a significant downregulation of DYRK2 in CD34+ cells from patients in the chronic phase (Figure 3F). We further performed quantitative polymerase chain reaction (qPCR) analysis of purified subsets and showed that DYRK2 transcripts were reduced in stem (CD34+ CD38−) and progenitor (CD34+ CD38+) cells from CML patient samples compared with those in corresponding cells from healthy donors (Figure 3G). Finally, a panel of leukemic cell lines revealed low DYRK2 levels in the CML cell lines K562, KU-812, and KBM5 (Figure 3H). These findings strengthen the model of DYRK2 repression as a key event in the maintenance of CML; however, the variability of DYRK2 messenger RNA levels seen in CML patients suggests the presence of additional posttranscriptional mechanisms to prevent DYRK2 inhibition in LSCs.
DYRK2 promotes c-Myc degradation and p53 activation in CML stem/progenitor cells
DYRK2 has been described to induce proteasomal degradation of c-Myc via prime phosphorylation at Ser 62 and apoptosis via p53 phosphorylation at Ser 46 in cancer cell lines HeLa and U2OS.35,36 To investigate whether DYRK2 also regulates p53 and c-Myc in CML, we purified GFP+ LSK cells from CML mice by cell sorting and analyzed them by immunoblotting. The upregulation of DYRK2 in Klf4Δ/Δ GFP+ LSK cells correlated with depletion of c-Myc protein and elevated levels of phosphorylated p53 (Ser 46), whereas the phosphorylated c-Myc (S62) protein was undetectable (Figure 4A). The lack of c-Myc phosphorylated at Ser 62 in Klf4Δ/Δ LSCs suggests that c-Myc is not stabilized in CML stem/progenitor cells by Ser 62 phosphorylation, as reported in breast cancer downstream of the Ras/ERK pathway.40 Consistent with this finding, gene set enrichment analysis showed higher expression of genes regulated by p53 and apoptosis, whereas genes downstream of c-Myc were downregulated (supplemental Figure 6). Genomic silencing of KLF4 in the cell line 32D-BCR-ABL1 resulted in DYRK2 upregulation and reduced c-Myc protein without alterations in GSK3 expression, which phosphorylates c-Myc at Thr 58 (Figure 4B).35 To further investigate whether c-Myc was posttranslationally regulated, we found that inhibition of proteolysis resulted in increased levels of total c-Myc, as well as in detectable levels of c-Myc phosphorylated at Ser 62 and Thr 58, which are the sites of DYRK2 and GSK3 phosphorylation, respectively (Figure 4C). Our findings uncovered a novel regulation of c-Myc in CML via DYRK2 phosphorylation and suggest that DYRK2 emerges as a key regulator of p53 and c-Myc, which are known to play key roles in CML.41,42
![DYRK2 inhibits self-renewal in leukemia stem/progenitor cells by inducing c-Myc degradation. (A) Immunoblot analysis of DYRK2, c-MYC, phosphorylated c-MYC (Ser 62) [P-c-Myc (S62)] and phosphorylated p53 (Ser 46) [P-p53 (S46)] expression in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. Actin was used as a loading control. (B) Immunoblot analysis of KLF4, DYRK2, c-MYC, and GSK3α expression in the 32D-BCR-ABL1 cell line transduced with short hairpin RNA (shRNA) retrovirus specific for luciferase (Luc) or KLF4. Two different KLF4 shRNAs are shown. (C) Effect of proteasome inhibition with MG-132 in the 32D-BCR-ABL1 cell line transduced with luciferase or KLF4 shRNA retrovirus (sh2) on the expression of c-MYC (total and phosphorylated at Ser 62 or at Thr 58). (D) Analysis of endogenous c-MYC levels in leukemia stem/progenitor cells from CML mice 14 days after transplantation of Klf4Δ/Δc-Mycgfp/gfp or Klf4fl/flc-Mycgfp/gfp bone marrow cells transduced with BCR-ABL1-RFP retrovirus. c-Mycgfp/gfp mice express the c-Myc–GFP fusion protein knocked in the c-Myc locus to monitor the expression of c-MYC (GFP) by flow cytometry in RFP(BCR-ABL1)+ LSK cells. The percentage of RFP+ LSK cells with high c-Myc–GFP is also shown (right panel; n = 5, mean). (E) Survival of mice transplanted with Klf4Δ/Δ Lin− Sca-l+ cells cotransduced with BCR-ABL1-GFP and empty vector–RFP (n = 5) or BCR-ABL1-GFP and c-MYCS62A mutant–RFP (n = 10). (F) Gr1 expression in leukemic cells (GFP+) was determined in peripheral blood at 24 dpt. (G) Upregulation of DYRK2 and c-Myc depletion in K562 cells lacking KLF4. (H) Diagram shows the regulation of p53 and c-Myc by DYRK2 downstream of KLF4 and the effect of loss of KLF4. The data are representative of 2 or 3 independent experiments. **P < .01, 2-tailed Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2018875922/3/m_blood875922f4.png?Expires=1769470553&Signature=PKlD0kGiwkak4zDNoSTq~guXaa5hfKWkED5IY~z8RLHXrkZ2OPwOKwzpS5rVo6TUVgmcHIw1~~LFoJKrxOedBVOWcebieFHMFuySA4oEtJtXz6v5s3kJ9IdiyAwsTupzZqZdFM5KcF5Vf5RNOclkHnV65XIg-Bb6QBWY~t0QoVtXKmIVdCfuK29yfK~asRgYuMxFuknrAKWMyKEKWOIZzH4IqnTVrlwZ586qgIhfvnGbHRP0WdVPI0yDltUj1Qtdg6M9pz06AqQ9NzP-RtWQ4CYQ6g8mCaTmZhKTIY7u4BjUhUL1PI5PnFWPIKiOpKIvGrhSAzzDI6f9LpxWNXmugw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
DYRK2 inhibits self-renewal in leukemia stem/progenitor cells by inducing c-Myc degradation. (A) Immunoblot analysis of DYRK2, c-MYC, phosphorylated c-MYC (Ser 62) [P-c-Myc (S62)] and phosphorylated p53 (Ser 46) [P-p53 (S46)] expression in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. Actin was used as a loading control. (B) Immunoblot analysis of KLF4, DYRK2, c-MYC, and GSK3α expression in the 32D-BCR-ABL1 cell line transduced with short hairpin RNA (shRNA) retrovirus specific for luciferase (Luc) or KLF4. Two different KLF4 shRNAs are shown. (C) Effect of proteasome inhibition with MG-132 in the 32D-BCR-ABL1 cell line transduced with luciferase or KLF4 shRNA retrovirus (sh2) on the expression of c-MYC (total and phosphorylated at Ser 62 or at Thr 58). (D) Analysis of endogenous c-MYC levels in leukemia stem/progenitor cells from CML mice 14 days after transplantation of Klf4Δ/Δc-Mycgfp/gfp or Klf4fl/flc-Mycgfp/gfp bone marrow cells transduced with BCR-ABL1-RFP retrovirus. c-Mycgfp/gfp mice express the c-Myc–GFP fusion protein knocked in the c-Myc locus to monitor the expression of c-MYC (GFP) by flow cytometry in RFP(BCR-ABL1)+ LSK cells. The percentage of RFP+ LSK cells with high c-Myc–GFP is also shown (right panel; n = 5, mean). (E) Survival of mice transplanted with Klf4Δ/Δ Lin− Sca-l+ cells cotransduced with BCR-ABL1-GFP and empty vector–RFP (n = 5) or BCR-ABL1-GFP and c-MYCS62A mutant–RFP (n = 10). (F) Gr1 expression in leukemic cells (GFP+) was determined in peripheral blood at 24 dpt. (G) Upregulation of DYRK2 and c-Myc depletion in K562 cells lacking KLF4. (H) Diagram shows the regulation of p53 and c-Myc by DYRK2 downstream of KLF4 and the effect of loss of KLF4. The data are representative of 2 or 3 independent experiments. **P < .01, 2-tailed Student t test.
DYRK2 inhibits self-renewal in leukemia stem/progenitor cells by inducing c-Myc degradation. (A) Immunoblot analysis of DYRK2, c-MYC, phosphorylated c-MYC (Ser 62) [P-c-Myc (S62)] and phosphorylated p53 (Ser 46) [P-p53 (S46)] expression in GFP+ LSK cells purified from Klf4fl/fl and Klf4Δ/Δ CML mice. Actin was used as a loading control. (B) Immunoblot analysis of KLF4, DYRK2, c-MYC, and GSK3α expression in the 32D-BCR-ABL1 cell line transduced with short hairpin RNA (shRNA) retrovirus specific for luciferase (Luc) or KLF4. Two different KLF4 shRNAs are shown. (C) Effect of proteasome inhibition with MG-132 in the 32D-BCR-ABL1 cell line transduced with luciferase or KLF4 shRNA retrovirus (sh2) on the expression of c-MYC (total and phosphorylated at Ser 62 or at Thr 58). (D) Analysis of endogenous c-MYC levels in leukemia stem/progenitor cells from CML mice 14 days after transplantation of Klf4Δ/Δc-Mycgfp/gfp or Klf4fl/flc-Mycgfp/gfp bone marrow cells transduced with BCR-ABL1-RFP retrovirus. c-Mycgfp/gfp mice express the c-Myc–GFP fusion protein knocked in the c-Myc locus to monitor the expression of c-MYC (GFP) by flow cytometry in RFP(BCR-ABL1)+ LSK cells. The percentage of RFP+ LSK cells with high c-Myc–GFP is also shown (right panel; n = 5, mean). (E) Survival of mice transplanted with Klf4Δ/Δ Lin− Sca-l+ cells cotransduced with BCR-ABL1-GFP and empty vector–RFP (n = 5) or BCR-ABL1-GFP and c-MYCS62A mutant–RFP (n = 10). (F) Gr1 expression in leukemic cells (GFP+) was determined in peripheral blood at 24 dpt. (G) Upregulation of DYRK2 and c-Myc depletion in K562 cells lacking KLF4. (H) Diagram shows the regulation of p53 and c-Myc by DYRK2 downstream of KLF4 and the effect of loss of KLF4. The data are representative of 2 or 3 independent experiments. **P < .01, 2-tailed Student t test.
To further investigate the regulation of c-Myc and self-renewal in leukemic stem/progenitor cells, we crossed Klf4fl/fl and Klf4Δ/Δ mice with mice carrying a Myc:GFP fusion knock-in at the endogenous Myc locus to determine, in vivo, the c-Myc levels in leukemic stem/progenitor cells generated through the use of a retrovirus carrying BCR-ABL1-RFP.30,43,44 Flow cytometric detection of GFP, as a marker of endogenous c-Myc expression, showed significant downregulation in Klf4Δ/Δ RFP+ LSK cells compared with that in control RFP+ LSK cells (Figure 4D), supporting an important role for KLF4 in the maintenance of c-Myc in CML stem/progenitor cells. Conversely, c-Myc levels in normal hematopoietic stem/progenitor cells (Klf4Δ/Δ RFP− LSK) were similar to those in control cells (supplemental Figure 7). To confirm that DYRK2-mediated degradation of c-Myc was required to abolish LSC self-renewal and CML progression, we cotransduced Klf4Δ/Δ HSCs with retroviruses carrying BCR-ABL1-IRES-GFP, c-MycS62A-IRES-RFP, or empty-IRES-RFP (control). The c-MycS62A mutation is a single amino acid substitution mutation at the DYRK2 phosphorylation site (S62A) in c-Myc that prevents DYRK2 phosphorylation and subsequent proteolysis by the proteasome. Mice transplanted with RFP+ and GFP+ cells were monitored for disease progression. Most mice transplanted with transformed Klf4Δ/Δ HSCs survived, whereas retroviral expression of the c-MycS62A mutant in Klf4Δ/Δ leukemic cells restored expansion of myeloid leukemia cells and development of myeloid leukemia that resulted in the demise of all mice (Figure 4E-F). To correlate our observations in murine leukemia with human CML cells, we knocked out the KLF4 gene in K562 cells through CRISPR/CAS9 and performed immunoblot analysis that showed similar DYRK2 upregulation and reduced c-MYC expression upon KLF4 silencing (Figure 4G). Altogether, our data support the model of KLF4 inhibition of the DYRK2-mediated activation of p53 and c-Myc degradation in BCR-ABL1+ LSK cells; thus, DYRK2 emerges as a single target with a dual function, self-renewal and survival, in the maintenance of leukemic stem/progenitor cells (Figure 4H).
Antileukemic properties of DYRK2 stabilization through inhibition of proteasomal degradation in human CML cells
Pharmacological activation or stabilization of the DYRK2 protein could represent a novel therapeutic approach to efficiently eradicate leukemia-initiating cells in CML patients. Based on 2 reports showing that the ubiquitin ligase SIAH2 mediates proteasomal degradation of DYRK2 and that the chemical compound menadione inhibits SIAH2,45-47 we propose that menadione, also known as VK3, could induce apoptosis and abrogate self-renewal in CML cells by stabilizing the DYRK2 protein. Treatment of the CML cell lines K562, KCL-22, and KU-812 with VK3 revealed dose-dependent cytotoxicity (Figure 5A) that was associated with increased levels of DYRK2, cleavage of caspase 3 and PARP, and depletion of c-Myc protein (Figure 5B). In contrast to VK3, natural vitamins K1 and K2 were unable to induce cytotoxicity in K562 cells, suggesting that the isoprenoid side chain may hinder binding and inhibition of SIAH2 (supplemental Figure 8). Menadiol, a reduced form of VK3, showed similar cytotoxicity, suggesting that antileukemic properties are likely independent of oxidative capacity (supplemental Figure 9). In terms of the specificity of SIAH2 inhibition by VK3, the lack of expression of SIAH1 in K562 and KCL-22 cells and upregulation of PHD3, a known target of SIAH2, by VK3 in all CML cell lines (Figure 5B) suggest that cytotoxicity of VK3 was primarily mediated by inhibition of SIAH2.48 Several pieces of evidence support that VK3 cytotoxicity is mediated through DYRK2 upregulation: the effect of VK3 on DYRK2 upregulation, c-Myc depletion, and PARP cleavage was prevented by knocking out DYRK2 in K562 cells via CRISPR/CAS9-mediated genome editing (Figure 5C), VK3 significantly reduced the ubiquitination of DYRK2 in an immunoprecipitation assay in K562 cells (Figure 5D), and sensitivity to VK3 was significantly reduced in K562 cells with the DYRK2 gene knocked out particularly at concentrations that induce DYRK2 expression (Figure 5E; supplemental Figure 10). Although the induction of apoptosis by VK3 in CML cell lines lacking p53 indicates that c-Myc depletion may be the main cause of cytotoxicity,42,49 we further investigated the role of p53 in VK3-mediated cytotoxicity. Treatment of K562 cells overexpressing p53 with VK3 resulted in increased sensitivity in cytotoxicity assays (data not shown) and an increased percentage of annexin V+ cells in a dose-dependent manner (Figure 5F).
![Antileukemic properties of DYRK2 stabilization with VK3. (A) Cell viability of KU-812, K562, and KCL-22 CML cells that were incubated for 48 hours in the presence of VK3 expressed as a percentage of the vehicle control (mean ± standard deviation [SD]). (B) Immunoblot analysis of DYRK2, c-Myc, SIAH1, SIAH2, and PHD3, as well as cleavage of PARP (cPARP) and caspase 3 (cCASP3), in protein lysates of CML cell lines treated with vehicle or 20 μM VK3. (C) Levels of DYRK2, c-Myc, and cleaved PARP (cPARP) in CRISPR/CAS9-mediated DYRK2-knockout K562 cells treated with vehicle or 12 μM VK3 for 48 hours. (D) Effect of VK3 on DYRK2 ubiquitination was analyzed in K562 cells treated with MG-132 and VK3 by immunoprecipitation with anti-DYRK2, and immunoblots were revealed with anti-ubiquitin. DYRK2 level is shown in protein lysates. (E) VK3 cytotoxicity in K562 cells with the DYRK2 gene knocked out by CRISPR/CAS9 compared with that in the parental cell line (n = 3, mean ± SD) (left panel) and immunoblotting of DYRK2 gene deletion (right panel). (F) Dose-dependent induction of apoptosis in K562 cells with p53 overexpression. (G) Effect of the combination of VK3 and IM on the viability of K562 cells (mean ± SD). (H) Isobologram analysis of VK3 and IM combination. (I) Cytotoxicity of VK3 in K562 cells resistant to IM (IM R), generated by culture in the presence of 2.5 μM IM, compared with that in parental cells (mean ± SD). (J) Cell viability (left panel) and immunoblots (right panel) of K562 cells incubated with menadione (VK3) or VK3-BS (mean ± SD). DYRK2 upregulation is shown (right panel). The data are representative of 2 or 3 independent experiments. *P < .05, ***P < .001, ****P < .0001, 2-tailed Student t test. Fa, fraction affected.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2018875922/3/m_blood875922f5.png?Expires=1769470553&Signature=FjeuX-h9TKm9u1JOaY7EMnTLWM88kB6Ph-UMmf2KX6mzYGCyJ069qmE2pTyeWwgNehniW5yIG1dtpgvZrJdpjuadjuD1ox~jHQ0JuxshYLxkaRtPbMdHuCEs16q~pQix~WdkkMXfJhOK6OinOipf2D3KyGOorudiDUP7~ao8MZXhbpf33lZW69np5BjWJkPIrlbAfFvpC~ePyy~Ld5ll8IQA5Jo2OizbWK5K9HT8su6kzlypYSUbkBKqK8kpAYsKyIYlx06tgeoVoSiiphvO7kYrtMr4UNBH3qlNmpmA3MKsllD9eM~PASMnHG7jsSh4aX-PoZpEpAGh5a1eJzJ4wg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Antileukemic properties of DYRK2 stabilization with VK3. (A) Cell viability of KU-812, K562, and KCL-22 CML cells that were incubated for 48 hours in the presence of VK3 expressed as a percentage of the vehicle control (mean ± standard deviation [SD]). (B) Immunoblot analysis of DYRK2, c-Myc, SIAH1, SIAH2, and PHD3, as well as cleavage of PARP (cPARP) and caspase 3 (cCASP3), in protein lysates of CML cell lines treated with vehicle or 20 μM VK3. (C) Levels of DYRK2, c-Myc, and cleaved PARP (cPARP) in CRISPR/CAS9-mediated DYRK2-knockout K562 cells treated with vehicle or 12 μM VK3 for 48 hours. (D) Effect of VK3 on DYRK2 ubiquitination was analyzed in K562 cells treated with MG-132 and VK3 by immunoprecipitation with anti-DYRK2, and immunoblots were revealed with anti-ubiquitin. DYRK2 level is shown in protein lysates. (E) VK3 cytotoxicity in K562 cells with the DYRK2 gene knocked out by CRISPR/CAS9 compared with that in the parental cell line (n = 3, mean ± SD) (left panel) and immunoblotting of DYRK2 gene deletion (right panel). (F) Dose-dependent induction of apoptosis in K562 cells with p53 overexpression. (G) Effect of the combination of VK3 and IM on the viability of K562 cells (mean ± SD). (H) Isobologram analysis of VK3 and IM combination. (I) Cytotoxicity of VK3 in K562 cells resistant to IM (IM R), generated by culture in the presence of 2.5 μM IM, compared with that in parental cells (mean ± SD). (J) Cell viability (left panel) and immunoblots (right panel) of K562 cells incubated with menadione (VK3) or VK3-BS (mean ± SD). DYRK2 upregulation is shown (right panel). The data are representative of 2 or 3 independent experiments. *P < .05, ***P < .001, ****P < .0001, 2-tailed Student t test. Fa, fraction affected.
Antileukemic properties of DYRK2 stabilization with VK3. (A) Cell viability of KU-812, K562, and KCL-22 CML cells that were incubated for 48 hours in the presence of VK3 expressed as a percentage of the vehicle control (mean ± standard deviation [SD]). (B) Immunoblot analysis of DYRK2, c-Myc, SIAH1, SIAH2, and PHD3, as well as cleavage of PARP (cPARP) and caspase 3 (cCASP3), in protein lysates of CML cell lines treated with vehicle or 20 μM VK3. (C) Levels of DYRK2, c-Myc, and cleaved PARP (cPARP) in CRISPR/CAS9-mediated DYRK2-knockout K562 cells treated with vehicle or 12 μM VK3 for 48 hours. (D) Effect of VK3 on DYRK2 ubiquitination was analyzed in K562 cells treated with MG-132 and VK3 by immunoprecipitation with anti-DYRK2, and immunoblots were revealed with anti-ubiquitin. DYRK2 level is shown in protein lysates. (E) VK3 cytotoxicity in K562 cells with the DYRK2 gene knocked out by CRISPR/CAS9 compared with that in the parental cell line (n = 3, mean ± SD) (left panel) and immunoblotting of DYRK2 gene deletion (right panel). (F) Dose-dependent induction of apoptosis in K562 cells with p53 overexpression. (G) Effect of the combination of VK3 and IM on the viability of K562 cells (mean ± SD). (H) Isobologram analysis of VK3 and IM combination. (I) Cytotoxicity of VK3 in K562 cells resistant to IM (IM R), generated by culture in the presence of 2.5 μM IM, compared with that in parental cells (mean ± SD). (J) Cell viability (left panel) and immunoblots (right panel) of K562 cells incubated with menadione (VK3) or VK3-BS (mean ± SD). DYRK2 upregulation is shown (right panel). The data are representative of 2 or 3 independent experiments. *P < .05, ***P < .001, ****P < .0001, 2-tailed Student t test. Fa, fraction affected.
The ideal therapy to achieve treatment-free remission would be a combination of a TKI with an as yet unidentified LSC-specific drug. Therefore, we studied the effects of VK3 and imatinib mesylate (IM) and found that this combination induced cytotoxicity in K562 cells at lower doses compared with single drugs, and an analysis of the drug combination revealed a synergistic effect (Figure 5G-H). In addition, VK3 inhibited the viability of IM-resistant K562 cells generated by incubating K562 cells with IM (2.5 μM) for 2 weeks (Figure 5I; supplemental Figure 11).50 Finally, we evaluated whether the hydrophilic compound VK3 sodium bisulfite (VK3-BS) shows cytotoxicity and upregulation of DYRK2 protein similar to those of liposoluble VK3 for in vivo studies. A similar cytotoxicity for both VK3 compounds and upregulation of DYRK2 suggest that VK3-BS can be used in mice with CML-like leukemia instead of VK3 to reduce the toxicity of the vehicle (Figure 5J).
DYRK2 stabilization targets leukemia stem/progenitor cells in mouse and human CML models
To treat mice with CML-like myeloproliferative neoplasia and patient-derived xenografts, we used VK3-BS. Analysis of VK3-BS toxicity in wild-type mice did not reveal any gross alterations in body weight or blood lineages with the exception of slightly reduced red blood cell numbers (supplemental Figure 12). In addition, the frequency of bone marrow HSCs, defined as LSK CD150+ CD48− cells, was unaffected by VK3-BS administration (Figure 6A). In the CML-like model, in vitro culture of purified GFP+ LSK cells, a population enriched in leukemic stem/progenitor cells, with VK3 resulted in increased levels of DYRK2 protein (Figure 6B). Next, we treated BCR-ABL1–induced CML mice with daily VK3-BS administration for 14 days to evaluate its efficacy in eliminating LSCs (Figure 6B). The administration of VK3-BS to mice with primary CML-like disease significantly diminished the frequency of bulk leukemic cells (GFP+ Gr1+) and leukemia stem/progenitor cells (GFP+ LSK cells) in bone marrow; most importantly, it reduced the leukemia burden and improved overall survival in secondary recipients, suggesting efficient elimination of leukemic stem/progenitor cells with leukemia-initiating capacity via systemic VK3-BS administration (Figure 6C-D; supplemental Figure 13). To correlate our findings with human disease, we next tested whether DYRK2 stabilization could induce apoptosis and inhibit self-renewal in bone marrow cells from chronic-phase CML patients. Annexin V staining showed increased cell death in bone marrow cells from patients with CML that were incubated in vitro with VK3 that was not seen in bone marrow cells from healthy individuals (Figure 6E; supplemental Figure 14). This cytotoxicity was associated with increased levels of DYRK2 and p53 phosphorylated at Ser 46, whereas c-Myc depletion was not distinguishable because of the low c-Myc levels in whole bone marrow (Figure 6F). Strikingly, VK3 abrogated the capacity of CD34+ CML cells to self-renew and generate colonies in methylcellulose (Figure 6G; supplemental Figure 15). In another model, daily administration of VK3-BS significantly inhibited the growth of subcutaneous tumors induced by implanting K562 cells embedded in Matrigel into the flank of nude mice, which was confirmed by analysis of gross morphology, tumor weight, DYRK2 expression, and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling staining in excised tumors (supplemental Figure 16). Finally, we evaluated the effect of VK3-BS on human CD34+ CML cells using a xenograft model. CD34+ cells were purified by cell sorting from cryopreserved bone marrow cells from chronic-phase patients collected at diagnosis and then transplanted into sublethally irradiated NSG mice (5 × 104 CD34+ cells per mouse) (Figure 6H). Four weeks after xenotransplantation, NSG mice with similar engraftment were randomized for treatment with vehicle or VK3-BS for 2 weeks. Flow cytometric detection of CD45+ human leukemic blasts and detection of BCR-ABL transcripts by qPCR in bone marrow at the end of treatment showed that, compared with mice treated with the vehicle control, mice treated with VK3-BS exhibited a significant reduction in the frequency of human leukemia cells in peripheral blood and undetectable BCR-ABL in bone marrow (Figure 6I; supplemental Figure 17). These data showed that VK3 inhibits the survival of CML cells in a model of human CML and validated our findings in the CML mouse model. Notably, although we used VK3 as proof of concept of DYRK2 stabilization in CML stem/progenitor cells, clinical translation will require the identification of more specific SIAH2 inhibitors with lower hematological toxicity, evaluation of the safety of SIAH2 inhibition, and/or development of alternative approaches to activate DYRK2 in CML LSCs.
![VK3-BS targets murine and human CML stem/progenitor cells through DYRK2. (A) Analysis of HSCs in bone marrow of wild-type mice after 2 weeks of daily administration of VK3-BS. (B) GFP+ LSK cells were isolated from mice 8 dpt with BCR-ABL1–transduced HSCs to confirm DYRK2 upregulation upon in vitro incubation with VK3. CML mice were treated daily for 2 weeks with VK3-BS or vehicle starting at 8 days posttransplant (arrows). After treatment, bone marrow cells were analyzed by flow cytometry and transplanted into secondary mice to evaluate residual leukemia stem/progenitor cells with leukemia-initiating capacity. (C) Detection of myeloid leukemia cells and frequency of GFP+ LSK cells in whole bone marrow of primary CML at the end of treatment (n = 8, mean ± standard deviation [SD]). (D) Frequency of GFP+ Gr1+ leukemic cells at 24 dpt and overall survival in secondary CML. (E) Annexin V staining of bone marrow cells from chronic-phase CML patients (Pt) and healthy individuals incubated in vitro with vehicle or VK3 for 48 hours (triplicates, mean ± SD). (F) Stabilization of DYRK2 protein leading to the phosphorylation of p53-S46 and c-Myc reduction in bone marrow from 2 CML patients by treatment with VK3 compared with those in bone marrow cells from a healthy donor. (G) Colony formation in methylcellulose media of CD34+ cells from chronic-phase CML patients and normal individuals treated with VK3 for 24 hours (triplicates, mean ± SD) (left panel). Representative images of colonies from CD34+ CML cells treated with 5 μM VK3 (right panels) and control. (H) Experimental design for the CML xenograft model: CD34+ stem/progenitor cells from CML patients were transplanted into sublethally irradiated NSG mice (5 × 104 cells per mouse); engraftment was confirmed by flow cytometry after 4 weeks, and mice were randomized for daily treatment (arrows) with vehicle or VK3 for 2 weeks. (I) Detection of human CD45 blood cells (hCD45) vs murine CD45 blood cells (mCD45) was determined by flow cytometry in blood and qPCR detection of BCR-ABL in bone marrow at the end of treatment (see also supplemental Figure 17). The data are representative of ≥3 independent experiments. *P < .05,**P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test and the log-rank test. BMT, bone marrow transplantation; n.d., not detected; n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2018875922/3/m_blood875922f6.png?Expires=1769470553&Signature=L~bR-iV3rcfjey41YyMyRMRJ5f8O2d3Qgli-lOWY40QkuWYfDtTdfafnDr8HOCa98AhhKXZoe--s68sX38aZOJ4hs6Fnmdh6q-lQmJmiy~j8X9n3aLI4BkBrw6~kIYncDmdnOPQZa4NOSvomoXjrn6Ow3bGzFjY6fqnBHnQPIPOyvUkd9O77QO4nCc2hO9R4~VtY0B5NiKFDSmPr1OU4fkwjhuFS1vn1apy5-G43quLNlpceeXpdC66Sf-JW6yMcoKN6p6Mt6H1LumTnZ1WtOfSEdKfJO1Is4FL5IN9G7JMdDGVM~txBLy3Pa4TYRDVFZaIPhcO6235rVkLmzdKqTw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
VK3-BS targets murine and human CML stem/progenitor cells through DYRK2. (A) Analysis of HSCs in bone marrow of wild-type mice after 2 weeks of daily administration of VK3-BS. (B) GFP+ LSK cells were isolated from mice 8 dpt with BCR-ABL1–transduced HSCs to confirm DYRK2 upregulation upon in vitro incubation with VK3. CML mice were treated daily for 2 weeks with VK3-BS or vehicle starting at 8 days posttransplant (arrows). After treatment, bone marrow cells were analyzed by flow cytometry and transplanted into secondary mice to evaluate residual leukemia stem/progenitor cells with leukemia-initiating capacity. (C) Detection of myeloid leukemia cells and frequency of GFP+ LSK cells in whole bone marrow of primary CML at the end of treatment (n = 8, mean ± standard deviation [SD]). (D) Frequency of GFP+ Gr1+ leukemic cells at 24 dpt and overall survival in secondary CML. (E) Annexin V staining of bone marrow cells from chronic-phase CML patients (Pt) and healthy individuals incubated in vitro with vehicle or VK3 for 48 hours (triplicates, mean ± SD). (F) Stabilization of DYRK2 protein leading to the phosphorylation of p53-S46 and c-Myc reduction in bone marrow from 2 CML patients by treatment with VK3 compared with those in bone marrow cells from a healthy donor. (G) Colony formation in methylcellulose media of CD34+ cells from chronic-phase CML patients and normal individuals treated with VK3 for 24 hours (triplicates, mean ± SD) (left panel). Representative images of colonies from CD34+ CML cells treated with 5 μM VK3 (right panels) and control. (H) Experimental design for the CML xenograft model: CD34+ stem/progenitor cells from CML patients were transplanted into sublethally irradiated NSG mice (5 × 104 cells per mouse); engraftment was confirmed by flow cytometry after 4 weeks, and mice were randomized for daily treatment (arrows) with vehicle or VK3 for 2 weeks. (I) Detection of human CD45 blood cells (hCD45) vs murine CD45 blood cells (mCD45) was determined by flow cytometry in blood and qPCR detection of BCR-ABL in bone marrow at the end of treatment (see also supplemental Figure 17). The data are representative of ≥3 independent experiments. *P < .05,**P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test and the log-rank test. BMT, bone marrow transplantation; n.d., not detected; n.s., not significant.
VK3-BS targets murine and human CML stem/progenitor cells through DYRK2. (A) Analysis of HSCs in bone marrow of wild-type mice after 2 weeks of daily administration of VK3-BS. (B) GFP+ LSK cells were isolated from mice 8 dpt with BCR-ABL1–transduced HSCs to confirm DYRK2 upregulation upon in vitro incubation with VK3. CML mice were treated daily for 2 weeks with VK3-BS or vehicle starting at 8 days posttransplant (arrows). After treatment, bone marrow cells were analyzed by flow cytometry and transplanted into secondary mice to evaluate residual leukemia stem/progenitor cells with leukemia-initiating capacity. (C) Detection of myeloid leukemia cells and frequency of GFP+ LSK cells in whole bone marrow of primary CML at the end of treatment (n = 8, mean ± standard deviation [SD]). (D) Frequency of GFP+ Gr1+ leukemic cells at 24 dpt and overall survival in secondary CML. (E) Annexin V staining of bone marrow cells from chronic-phase CML patients (Pt) and healthy individuals incubated in vitro with vehicle or VK3 for 48 hours (triplicates, mean ± SD). (F) Stabilization of DYRK2 protein leading to the phosphorylation of p53-S46 and c-Myc reduction in bone marrow from 2 CML patients by treatment with VK3 compared with those in bone marrow cells from a healthy donor. (G) Colony formation in methylcellulose media of CD34+ cells from chronic-phase CML patients and normal individuals treated with VK3 for 24 hours (triplicates, mean ± SD) (left panel). Representative images of colonies from CD34+ CML cells treated with 5 μM VK3 (right panels) and control. (H) Experimental design for the CML xenograft model: CD34+ stem/progenitor cells from CML patients were transplanted into sublethally irradiated NSG mice (5 × 104 cells per mouse); engraftment was confirmed by flow cytometry after 4 weeks, and mice were randomized for daily treatment (arrows) with vehicle or VK3 for 2 weeks. (I) Detection of human CD45 blood cells (hCD45) vs murine CD45 blood cells (mCD45) was determined by flow cytometry in blood and qPCR detection of BCR-ABL in bone marrow at the end of treatment (see also supplemental Figure 17). The data are representative of ≥3 independent experiments. *P < .05,**P < .01, ***P < .001, ****P < .0001, 2-tailed Student t test and the log-rank test. BMT, bone marrow transplantation; n.d., not detected; n.s., not significant.
Discussion
The eradication of LSCs that are spared during standard chemotherapy and are able to self-renew and induce relapse constitutes a major challenge in the treatment of leukemia. This study identified the kinase DYRK2 as a critical checkpoint controlling self-renewal and survival in leukemia stem/progenitor cells in CML and showed that increasing levels of the DYRK2 protein, either by genetic loss of KLF4 or pharmacological inhibition of the ubiquitin ligase SIAH2, could induce apoptosis and abrogate self-renewal in CML stem/progenitor cells.
The reprogramming factor KLF4 plays critical roles in the control of self-renewal in normal and malignant stem cells, such as embryonic stem cells and leukemia initiating cells, in a cell context–dependent manner.22,30 KLF4 indirectly promotes self-renewal in BCR-ABL1+ leukemia stem/progenitor cells by repressing the DYRK2 gene via a mechanism complementary to the ubiquitin ligase SIAH2. There are several factors involved in the molecular control of self-renewal in chronic-phase CML LSCs, such as β-catenin,51 GABPβ transcription factor,52 Tcf1/Lef1 transcription factors,53 and adenosine deaminase ADAR1.54 A recent article describing the inhibition of stem cell self-renewal in CML by prostaglandin E1 included a gene expression profile in which KLF4, although not the focus of this study, strikingly appeared as 1 of the most downregulated genes, suggesting that the observed effect may be mediated by KLF4.55
In this study, DYRK2 emerged as a novel negative regulator of CML downstream of KLF4 by promoting apoptosis and inhibiting self-renewal through activation of p53 and degradation of c-Myc in leukemia stem/progenitor cells. Inhibitory functions have been ascribed to the kinase DYRK2 in breast cancer stem cells, paradoxically, via activation of the KLF4 gene downstream of androgen receptor signaling, as well as prevention of the formation of breast tumors mediated by proteasome activity by inducing degradation of the proteasome.56,57 Low DYRK2 levels have been associated with poor prognosis in colorectal cancer, recurrence in early-stage breast cancer, and proliferation in non-Hodgkin lymphoma.58-60 The downstream substrates of DYRK2, p53 and c-Myc, have been targeted simultaneously to eradicate LSCs and leukemic blasts in CML through stabilization of p53 with the MDM2 inhibitor RG7388 and inhibition of c-Myc expression with the BRET domain inhibitor CPI-203.41,42,61 Therefore, DYRK2 represents a single target, with a dual function in the activation of p53-mediated apoptosis and c-Myc proteolysis, that can potentially accelerate the process of LSC erosion during TKI treatment.62
The identification of DYRK2 as a key target to eliminate LSCs led us to investigate approaches that would mimic the effect of the genetic loss of KLF4, such as stabilization of DYRK2 protein by inhibiting proteasomal degradation. A study showed that CML patients exhibit elevated SIAH2 levels, and SIAH2 inhibition with VK3 attenuated chemoresistance in K562 cells.47 The ability of VK3 to enhance imatinib cytotoxicity and target imatinib-resistant CML cells suggests that the combination of SIAH2 inhibition with TKIs has the potential to simultaneously eliminate the bulk of leukemia cells and LSCs in CML patients by targeting different mechanisms. VK3 has been tested in clinical trials, in combination with mitomycin C, in lung cancer patients,63 in neonates to prevent vitamin K deficiency,64 in advanced hepatocellular carcinoma,65 and with the reduced form menadiol diphosphate in patients with advanced cancer.66 Although toxicities have been described for VK3 because of its capacity to generate reactive oxygen species,67,68 the use of VK3 and VK3-BS validates the model of DYRK2 as a master regulator of self-renewal and survival in leukemia stem/progenitor cells and supports the future development of LSC-specific therapy by inhibiting SIAH2 using the core naphthoquinone of VK3 as a lead compound, searching for more specific and less toxic inhibitors, or directly activating DYRK2 to avoid stabilization of other proteins potentially involved in CML.
Collectively, our study proposes a new model in which KLF4 represses the DYRK2 gene in CML stem/progenitor cells to prevent abrogation of self-renewal and survival. Because of these inhibitory functions, CML cells must suppress DYRK2 at 2 levels by repressing gene expression via KLF4 and inducing proteasomal degradation via the ubiquitin ligase SIAH2. Pharmacological inhibition of SIAH2 with VK3, as a means to increase DYRK2 protein, can target CML LSCs.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Tomasz Skorski (Temple University) for providing the 32D BCR-ABL1 cell line, W. Pear (University of Pennsylvania) for supplying the retroviral constructs, Bing Carter (University of Texas MD Anderson Cancer Center) for providing the KBM5 cell line, and Karen Prince for the preparation of figures.
This work was supported in part by the Rally Foundation for Childhood Cancer Research (H.D.L.), Gabrielle’s Angel Foundation for Cancer Research (H.D.L.), the Cancer Prevention Research Institute of Texas (RP140179) (H.D.L.), National Cancer Institute, National Institutes of Health (R01 CA207086-01A1) (H.D.L.), National Institute of General Medical Sciences T32, National Institutes of Health (GM008231) (T.C.), the Baylor College of Medicine Comprehensive Cancer Training Program from the Cancer Prevention and Research Institute of Texas (RP160283) (A.H.L.), the Cytometry and Cell Sorting Core at Baylor College of Medicine (P30 AI036211, P30 CA125123, and S10 RR024574), and the Flow Cytometry Core at Texas Children’s Cancer and Hematology Center (S10 OD020066).
Authorship
Contribution: C.S.P. designed and performed most of the experiments, interpreted the data, and wrote the manuscript; A.H.L. performed the cytotoxicity assays, serial replating of purified leukemic stem/progenitor cells, and ChIP-PCR; T.J.C. generated cell lines with genomic silencing and contributed to immunoblot analysis; C.S.B. contributed to the K562 xenograft model, cytotoxicity assays, and immunoblot analysis; Y.S. and K.S. contributed to generation of the CML mouse model; M.P. generated the different mouse strains used in this study; P.D.P. and J.A.T. performed VK3 cytotoxicity assays; R.R. contributed to the generation of DYRK2-knockout cell lines using Cas9/CRISPR technology; L.M. and M.R.G. performed the gene expression analyses in CML patients; T.-A.M. performed the bioinformatics analyses; and H.D.L. conceived the project, designed and interpreted the experiments, directed the project as the principal investigator, wrote the manuscript, and funded the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for K.S. is Center of Excellence for Cell and Stem Cell Therapy, King Chulalongkorn Memorial Hospital, Chulalongkorn University, Bangkok, Thailand.
Correspondence: H. Daniel Lacorazza, Baylor College of Medicine, Texas Children’s Hospital, 1102 Bates St FC830.20, Houston, TX 77030; e-mail: hdl@bcm.edu.